Packaging and storage—
Preserve in single-dose containers, preferably of Type I or Type II glass, and avoid excessive heat.
Labeling—
The label of the immediate container bears in a subtitle the name of the protein from which the hydrolysate has been derived and the word “modified” if one or more of the “essential” amino acids has been partially removed, restored, or added. The label bears a statement of the pH range; the name and percentage of any added other nutritive ingredient; the method of hydrolysis; the nature of the modification, if any, in amino acid content after hydrolysis; the percentage of each essential amino acid or its equivalent; the approximate protein equivalent, in g per liter; the approximate number of calories per liter; the percentage of the total nitrogen in the form of
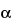
-amino nitrogen; and the quantity of the sodium and of the potassium ions present in each 100 mL of the Injection. Injection that contains not more than 30 mg of sodium per 100 mL may be labeled “Protein Hydrolysate Injection, Low Sodium,” or by a similar title the approximate equivalent thereof.
The label states the total osmolar concentration in mOsmol per L. Where the contents are less than 100 mL, or where the label states that the Injection is not for direct injection but is to be diluted before use, the label alternatively may state the total osmolar concentration in mOsmol per mL.
Non-antigenicity—
Sensitizing solution—
Select a suitable quantity of the protein identical in nature and quality with that from which the hydrolysate was manufactured, and subject it to the same hydrolytic process used in manufacturing the hydrolysate but reduce the time of hydrolysis to one-third. For purposes of preservation add, if desired, 0.5% of chlorobutanol, and package the sensitizing solution in 100-mL multiple-dose containers. Store in a cold place.
Preparation of animals—
Select healthy guinea pigs each weighing between 420 and 480 g. Inject each animal intraperitoneally with 6 mL of the sensitizing solution on the second, fourth, and sixth days of each of two successive weeks. Use the sensitized animals not less than 30 days and not more than 37 days after the last sensitizing dose. Re-sensitize any animals not used during the 7-day period by injecting intraperitoneally a booster dose of 6 mL of the sensitizing solution, and use re-sensitized animals not less than 9 days and not more than 16 days after the injection of the booster dose.
Procedure—
Inject, intravenously, 3 mL of Injection, at the rate of 2 mL per minute, using a 5-mL syringe fitted with a 27-gauge needle, into each of five guinea pigs prepared as described above. During the injection and during the 15 minutes following, observe the animals for any of the following symptoms: (1) licking the nose or rubbing the nose with forefeet; (2) ruffling of the fur; (3) labored breathing; (4) sneezing or coughing (three or more times); (5) retching. The requirements of the test are met if none of the injected animals show more than two of the listed symptoms and none show rales, convulsions, or prostration, or die. If none of the listed symptoms are observed, test the sensitivity of the animals by injecting into one of them 3 mL of the sensitizing solution intravenously at the rate of 2 mL per minute: the animal shows positive signs of anaphylaxis such as rales, convulsions, prostration, and/or death in addition to one or more of the lesser reaction symptoms.
pH  791
791 :
:
between 4.0 and 7.0, determined potentiometrically, but the variation from the pH range stated on the label is not greater than ±0.5 pH unit.
Content of -amino nitrogen—
Dilute 5.0 mL of a 5% Injection, or an appropriate volume of any other concentration, to 25 mL. Adjust to a pH of 7, potentiometrically, by the addition of 0.1 N sodium hydroxide or 0.1 N hydrochloric acid. Add 10 mL of formaldehyde TS, previously adjusted to a pH of 9.0, potentiometrically, then while stirring the solution, preferably with a mechanical stirrer, and with a suitable glass electrode in the system, add 0.1 N sodium hydroxide VS slowly toward the end, until a pH of 9.0 is reached. Continue stirring for 2 minutes, check the pH, and adjust if necessary. Record the volume of 0.1 N sodium hydroxide VS added in the titration. Each mL of 0.10 N sodium hydroxide corresponds to 1.4 mg of
-amino nitrogen.
Nitrogen content—
Using 0.1 mL of Injection, determine the nitrogen content as directed under
Method II (see
Nitrogen Determination 461).
Potassium content—
Standard solutions—
Prepare four standard solutions (numbered 1, 2, 3, and 4) each containing a suitable wetting agent and 25.0 mEq of sodium (1.46 g of sodium chloride) per liter, and to the solutions add, respectively, 0-, 2.0-, 3.0-, and 4.0-mg supplements of potassium, in the form of the chloride, per L. If necessary, because of changes in the sensitivity of the instrument mentioned below, vary the levels of concentration of the potassium, keeping the ratios between solutions approximately as given.
Standard graph—
Set a suitable flame photometer to a wavelength of 766 nm. Adjust the instrument to zero transmittance with solution 1. Then adjust the instrument to 100% transmittance with solution 4. Read the percent transmittance of solutions 2 and 3. Plot the observed transmittance of solutions 2, 3, and 4 as the ordinate and the concentration as the abscissa on arithmetic coordinate paper.
Procedure—
Pipet a portion of the Injection containing approximately 300 µg of potassium, or a quantity corresponding to the concentration of the Standard solutions, into a 100-mL volumetric flask. Add a small amount of wetting agent, and dilute to volume with a sodium solution of such strength that the final sodium concentration is 25.0 mEq per L. Adjust the instrument to zero transmittance with solution 1 and to 100% transmittance with solution 4. Read the percent transmittance of the test solution and, by reference to the standard potassium graph, calculate the potassium content of the Injection in mg of potassium per mL.
Sodium content—
Proceed as directed under
Potassium content, with the following modifications: (1) prepare the
Standard solutions to contain 6.00 mEq of potassium (447 mg of potassium chloride), and substitute sodium for the stated quantities of potassium; (2) prepare the
Standard graph with the flame photometer set at 589 nm instead of 766 nm; and (3) under
Procedure read “sodium” for “potassium” throughout, but in the second sentence read “a potassium solution of such strength that the final potassium concentration is 6.00 mEq per liter” in place of “a sodium solution of such strength that the final sodium concentration is 25.0 mEq per L.”
Other requirements—
It meets the requirements under
Injections 1.