Method Ia (Direct Titration)
Principle—
The titrimetric determination of water is based upon the quantitative reaction of water with an anhydrous solution of sulfur dioxide and iodine in the presence of a buffer that reacts with hydrogen ions.
In the original titrimetric solution, known as Karl Fischer Reagent, the sulfur dioxide and iodine are dissolved in pyridine and methanol. The test specimen may be titrated with the Reagent directly, or the analysis may be carried out by a residual titration procedure. The stoichiometry of the reaction is not exact, and the reproducibility of a determination depends upon such factors as the relative concentrations of the Reagent ingredients, the nature of the inert solvent used to dissolve the test specimen, and the technique used in the particular determination. Therefore, an empirically standardized technique is used in order to achieve the desired accuracy. Precision in the method is governed largely by the extent to which atmospheric moisture is excluded from the system. The titration of water is usually carried out with the use of anhydrous methanol as the solvent for the test specimen; however, other suitable solvents may be used for special or unusual test specimens.
Apparatus—
Any apparatus may be used that provides for adequate exclusion of atmospheric moisture and determination of the endpoint. In the case of a colorless solution that is titrated directly, the endpoint may be observed visually as a change in color from canary yellow to amber. The reverse is observed in the case of a test specimen that is titrated residually. More commonly, however, the endpoint is determined electrometrically with an apparatus employing a simple electrical circuit that serves to impress about 200 mV of applied potential between a pair of platinum electrodes immersed in the solution to be titrated. At the endpoint of the titration a slight excess of the reagent increases the flow of current to between 50 and 150 microamperes for 30 seconds to 30 minutes, depending upon the solution being titrated. The time is shortest for substances that dissolve in the reagent. With some automatic titrators, the abrupt change in current or potential at the endpoint serves to close a solenoid-operated valve that controls the buret delivering the titrant. Commercially available apparatus generally comprises a closed system consisting of one or two automatic burets and a tightly covered titration vessel fitted with the necessary electrodes and a magnetic stirrer. The air in the system is kept dry with a suitable desiccant, and the titration vessel may be purged by means of a stream of dry nitrogen or current of dry air.
Reagent—
Prepare the Karl Fischer Reagent as follows. Add 125 g of iodine to a solution containing 670 mL of methanol and 170 mL of pyridine, and cool. Place 100 mL of pyridine in a 250-mL graduated cylinder, and, keeping the pyridine cold in an ice bath, pass in dry sulfur dioxide until the volume reaches 200 mL. Slowly add this solution, with shaking, to the cooled iodine mixture. Shake to dissolve the iodine, transfer the solution to the apparatus, and allow the solution to stand overnight before standardizing. One mL of this solution when freshly prepared is equivalent to approximately 5 mg of water, but it deteriorates gradually; therefore, standardize it within 1 hour before use, or daily if in continuous use. Protect from light while in use. Store any bulk stock of the reagent in a suitably sealed, glass-stoppered container, fully protected from light, and under refrigeration.
A commercially available, stabilized solution of Karl Fischer type reagent may be used. Commercially available reagents containing solvents or bases other than pyridine or alcohols other than methanol may be used also. These may be single solutions or reagents formed in situ by combining the components of the reagents present in two discrete solutions. The diluted Reagent called for in some monographs should be diluted as directed by the manufacturer. Either methanol or other suitable solvent, such as ethylene glycol monomethyl ether, may be used as the diluent.
Test Preparation—
Unless otherwise specified in the individual monograph, use an accurately weighed or measured amount of the specimen under test estimated to contain 10 to 250 mg of water.
Where the specimen under test is an aerosol with propellant, store it in a freezer for not less than 2 hours, open the container, and test 10.0 mL of the well-mixed specimen. In titrating the specimen, determine the endpoint at a temperature of 10
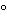
or higher.
Where the specimen under test is capsules, use a portion of the mixed contents of not fewer than 4 capsules.
Where the specimen under test is tablets, use powder from not fewer than 4 tablets ground to a fine powder in an atmosphere of temperature and relative humidity known not to influence the results.
Where the monograph specifies that the specimen under test is hygroscopic, use a dry syringe to inject an appropriate volume of methanol, or other suitable solvent, accurately measured, into a tared container, and shake to dissolve the specimen. Using the same syringe, remove the solution from the container and transfer it to a titration vessel prepared as directed for
Procedure. Repeat the procedure with a second portion of methanol, or other suitable solvent, accurately measured, add this washing to the titration vessel, and immediately titrate. Determine the water content, in mg, of a portion of solvent of the same total volume as that used to dissolve the specimen and to wash the container and syringe, as directed for
Standardization of Water Solution for Residual Titrations, and subtract this value from the water content, in mg, obtained in the titration of the specimen under test. Dry the container and its closure at 100
for 3 hours, allow to cool in a desiccator, and weigh. Determine the weight of specimen tested from the difference in weight from the initial weight of the container.
Standardization of the Reagent—
Place enough methanol or other suitable solvent in the titration vessel to cover the electrodes, and add sufficient
Reagent to give the characteristic endpoint color, or 100 ± 50 microamperes of direct current at about 200 mV of applied potential.
For determination of trace amounts of water (less than 1%), sodium tartrate may be used as a convenient water reference substance. Quickly add 150 to 350 mg of sodium tartrate (C4H4Na2O6·2H2O), accurately weighed by difference, and titrate to the endpoint. The water equivalence factor F, in mg of water per mL of reagent, is given by the formula:
2(18.02/230.08)(W/V),
in which 18.02 and 230.08 are the molecular weights of water and sodium tartrate dihydrate, respectively; W is the weight, in mg, of sodium tartrate dihydrate; and V is the volume, in mL, of the Reagent consumed in the second titration.
For the precise determination of significant amounts of water (1% or more), use
Purified Water as the reference substance. Quickly add between 25 and 250 mg of water, accurately weighed by difference, from a weighing pipet or from a precalibrated syringe or micropipet, the amount taken being governed by the reagent strength and the buret size, as referred to under
Volumetric Apparatus  31
31
. Titrate to the endpoint. Calculate the water equivalence factor,
F, in mg of water per mL of reagent, by the formula:
W/V,
in which W is the weight, in mg, of the water; and V is the volume, in mL, of the reagent required.
Procedure—
Unless otherwise specified, transfer 35 to 40 mL of methanol or other suitable solvent to the titration vessel, and titrate with the
Reagent to the electrometric or visual endpoint to consume any moisture that may be present. (Disregard the volume consumed, since it does not enter into the calculations.) Quickly add the
Test Preparation, mix, and again titrate with the
Reagent to the electrometric or visual endpoint. Calculate the water content of the specimen, in mg, taken by the formula:
SF,
in which
S is the volume, in mL, of the
Reagent consumed in the second titration; and
F is the water equivalence factor of the
Reagent.
Method Ib (Residual Titration)
Principle—
See the information given in the section Principle under Method Ia. In the residual titration, excess Reagent is added to the test specimen, sufficient time is allowed for the reaction to reach completion, and the unconsumed Reagent is titrated with a standard solution of water in a solvent such as methanol. The residual titration procedure is applicable generally and avoids the difficulties that may be encountered in the direct titration of substances from which the bound water is released slowly.
Apparatus, Reagent, and Test Preparation—
Use Method Ia.
Standardization of Water Solution for Residual Titration—
Prepare a
Water Solution by diluting 2 mL of water with methanol or other suitable solvent to 1000 mL. Standardize this solution by titrating 25.0 mL with the
Reagent, previously standardized as directed under
Standardization of the Reagent. Calculate the water content, in mg per mL, of the
Water Solution taken by the formula:
V¢F/25,
in which
V¢ is the volume of the
Reagent consumed, and
F is the water equivalence factor of the
Reagent. Determine the water content of the
Water Solution weekly, and standardize the
Reagent against it periodically as needed.
Procedure—
Where the individual monograph specifies that the water content is to be determined by
Method Ib, transfer 35 to 40 mL of methanol or other suitable solvent to the titration vessel, and titrate with the
Reagent to the electrometric or visual endpoint. Quickly add the
Test Preparation, mix, and add an accurately measured excess of the
Reagent. Allow sufficient time for the reaction to reach completion, and titrate the unconsumed
Reagent with standardized
Water Solution to the electrometric or visual endpoint. Calculate the water content of the specimen, in mg, taken by the formula:
F(
X¢ 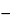 XR
XR),
in which
F is the water equivalence factor of the
Reagent; X¢ is the volume, in mL, of the
Reagent added after introduction of the specimen;
X is the volume, in mL, of standardized
Water Solution required to neutralize the unconsumed
Reagent; and
R is the ratio,
V¢/25 (mL
Reagent/mL
Water Solution), determined from the
Standardization of Water Solution for Residual Titration.
Method Ic (Coulometric Titration)
Principle—
The Karl Fischer reaction is used in the coulometric determination of water. Iodine, however, is not added in the form of a volumetric solution but is produced in an iodide-containing solution by anodic oxidation. The reaction cell usually consists of a large anode compartment and a small cathode compartment that are separated by a diaphragm. Other suitable types of reaction cells (e.g., without diaphragms) may also be used. Each compartment has a platinum electrode that conducts current through the cell. Iodine, which is produced at the anode electrode, immediately reacts with water present in the compartment. When all the water has been consumed, an excess of iodine occurs, which usually is detected electrometrically, thus indicating the endpoint. Moisture is eliminated from the system by pre-electrolysis. Changing the Karl Fischer solution after each determination is not necessary since individual determinations can be carried out in succession in the same reagent solution. A requirement for this method is that each component of the test specimen is compatible with the other components, and no side reactions take place. Samples are usually transferred into the vessel as solutions by means of injection through a septum. Gases can be introduced into the cell by means of a suitable gas inlet tube. Precision in the method is predominantly governed by the extent to which atmospheric moisture is excluded from the system; thus, the introduction of solids into the cell is not recommended, unless elaborate precautions are taken, such as working in a glove-box in an atmosphere of dry inert gas. Control of the system may be monitored by measuring the amount of baseline drift. This method is particularly suited to chemically inert substances like hydrocarbons, alcohols, and ethers. In comparison with the volumetric Karl Fischer titration, coulometry is a micro-method.
Apparatus—
Any commercially available apparatus consisting of an absolutely tight system fitted with the necessary electrodes and a magnetic stirrer is appropriate. The instrument's microprocessor controls the analytical procedure and displays the results. Calibration of the instrument is not necessary, as the current consumed can be measured absolutely.
Reagent—
See Reagent under Method Ia.
Test Preparation—
Where the specimen is a soluble solid, dissolve an appropriate quantity, accurately weighed, in anhydrous methanol or other suitable solvents. Liquids may be used as such or as accurately prepared solutions in appropriate anhydrous solvents.
Where the specimen is an insoluble solid, the water may be extracted using a suitable anhydrous solvent from which an appropriate quantity, accurately weighed, may be injected into the anolyte solution. Alternatively an evaporation technique may be used in which water is released and evaporated by heating the specimen in a tube in a stream of dry inert gas, this gas being then passed into the cell.
Procedure—
Using a dry syringe, quickly inject the Test Preparation, accurately measured and estimated to contain 0.5 to 5 mg of water, or as recommended by the instrument manufacturer into the anolyte, mix, and perform the coulometric titration to the electrometric endpoint. Read the water content of the Test Preparation directly from the instrument's display, and calculate the percentage that is present in the substance. Perform a blank determination, and make any necessary corrections.
;)