an ion source for producing gaseous ions from the substance being studied, an analyzer for resolving the ions into their characteristic mass components according to their mass-to-charge ratios, and a detector system for detecting the ions and recording the relative abundance of each of the resolved ionic species. In addition, a sample introduction system is necessary to admit the samples to be studied to the ion source while maintaining the high vacuum requirements (
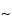
10
–6 to 10
–8 mm of mercury) of the technique; and a computer is required to control the instrument, acquire and manipulate data, and compare spectra to reference libraries.
This chapter gives an overview of the theory, construction, and use of mass spectrometers. The discussion is limited to those instruments and measurements with actual or potential application to compendial and other pharmaceutical requirements: generally, the identification and quantitation of specific compounds.
SAMPLE INTRODUCTION
Samples are introduced either as a gas to be ionized in the ion source, or by ejection of charged molecular species from a solid surface or solution. In some cases sample introduction and ionization take place in a single process, making a distinction between them somewhat artificial.
Substances that are gases or liquids at room temperature and atmospheric pressure can be admitted to the source as a neutral beam via a controllable leak system. Volatilizable compounds dissolved or adsorbed in solids or liquids can be removed and concentrated with a headspace analyzer. Vapors are flushed from the solid or liquid matrix with a stream of carrier gas and trapped on an adsorbing column. The trapped vapors are subsequently desorbed by programmed heating of the trap and introduced into the mass spectrometer by a capillary connection.
For volatilizable solids, the most frequently used method of sample introduction is the direct insertion probe. Here, the sample is placed in a small crucible at the tip of the probe, which is heated under high vacuum in close proximity to the ion source. A variation of this technique involves desorption of samples inside the ionization chamber from a rapidly heated wire or with the aid of a laser beam. Such desorption techniques, in combination with electron, chemical, or field ionization, are preferred for the analysis of heat sensitive or poorly volatile samples.
Sample introduction techniques that involve the ejection of charged molecules from the surface of solid samples include the field desorption method and various sputtering techniques, where the samples are bombarded by high energy photons, by a primary ion beam, or by a neutral particle beam. Similarly, ions can be ejected from solutions either by bombardment with a primary beam, or by one of the various spray techniques described below.
Gas and liquid chromatographs are widely used as sample inlet devices for mass spectrometers. These chromatographs provide for an initial sample purification, since only that portion of the chromatographic effluent containing the compound of interest need be admitted to the mass spectrometer. Gas chromatography/mass spectrometry (GC/MS) and liquid chromatography/mass spectrometry (LC/MS) combinations are valuable tools for the identification of unknown impurities in drugs. These combination methods have the capacity to separate complex mixtures with the opportunity to obtain structural information on the individual components.
Gas Chromatography/Mass Spectrometry
Gas chromatographic effluents are already in the vapor state and can be admitted directly into the mass spectrometer. Bridging the several orders of magnitude difference in the operating pressures of the two systems was initially accomplished with the use of various carrier gas separators. However, with the advent of capillary gas chromatographic columns and high capacity vacuum pumps for mass spectrometers, the gas chromatographic effluents are now fed directly into the ion source.
Liquid Chromatography/Mass Spectrometry
This technique is particularly useful for analyzing materials that cannot be analyzed by GC/MS, either because of thermal instability, high polarity, or high molecular weight. Compounds of biological interest such as drugs and their metabolites, polar endogenous substances, and macromolecules—including peptides, proteins, nucleic acids, and oligosaccharides—often fall into one of these categories.
Currently available LC/MS interfaces encompass a number of approaches to separating the compound of interest from the liquid chromatographic mobile phase and transforming it into an ionized species suitable for mass spectrometry. These include transport devices such as the particle beam; various spray techniques including thermospray, electrospray, and ionspray; and particle-induced desorption such as continuous-flow fast atom bombardment (CF-FAB).
PARTICLE BEAM INTERFACE
The solvent is removed from an aerosol of the liquid chromatographic effluent, and the resulting neutral analyte molecules are introduced into the ion source of the mass spectrometer where they are ionized by electron ionization (EI) or chemical ionization (CI). The resulting spectra are thus classical EI or CI spectra, the former with a wealth of structural information. There are limitations with respect to polarity, thermal lability, and molecular weight, so this technique is best suited for small organic molecules with molecular weights of less than 1000 daltons.
THERMOSPRAY
The compound of interest in a volatile buffer mobile phase, such as ammonium acetate, is passed through heated, narrow bore tubing directly into the ion source of a mass spectrometer. The solution is vaporized in the tubing, and analyte ions desorb into the gas phase and pass into the mass analyzer. Neutral analyte molecules in the gas phase may undergo chemical ionization by reaction with gas phase buffer ions such as NH4+. Thermospray is compatible with relatively high flow rates of 1 to 2 mL per minute, solvents containing a high percentage of water, and many types of polar analytes. Thermal degradation may occur, since the analytes are exposed to relatively high temperatures during the volatilization process.
ELECTROSPRAY
The mobile phase is sprayed through a small opening (needle tip) held at a potential of several kilovolts. The charged droplets so produced are desolvated by passing through a drying gas, and the resulting ions are injected directly into the high vacuum of the analyzer through an orifice or glass capillary. Classical electrospray is limited to flow rates of 1 to 5 µL per minute, and is therefore compatible with either microbore HPLC or post-column stream splitting techniques.
The ions may carry multiple charges, so that the m/z value of high molecular weight substances will fall into the usable range for most quadrupole or magnetic sector mass analyzers (m/z < 4000). Analytes of up to 150,000 daltons can thus be successfully analyzed.
IONSPRAY
A variant of electrospray in which nebulization with a gas flow is used to assist the formation of microdroplets of mobile phase. The technique can extend the upper limit of usable flow rates to 0.1 mL per minute. Volatile buffers must be used with both techniques.
DESORPTION TECHNIQUES
Microflow liquid chromatography can also be interfaced to particle induced desorption techniques such as fast atom bombardment (FAB) and liquid secondary ion mass spectroscopy (LSIMS), described in the following section on ionization techniques. Typically, column effluent flowing at a rate of 1 to 10 µL per minute is mixed with a small percentage of nonvolatile liquid such as glycerol. The mixture is introduced via a capillary inlet onto a target within the ion source where it is bombarded with high energy (5 to 20 keV) atoms or ions. The resulting spectra are similar to FAB or LSIMS spectra but with the background from the sample matrix greatly reduced. Frit-FAB is a variant of continuous flow FAB where the sample is introduced through a porous frit target.
ANALYZERS
Mass analyzers separate the charged species in the ionized sample according to their m/z ratios, thus permitting the mass and abundance of each species to be determined. Four commonly used analyzers are the magnetic sector, the quadrupole, the time-of-flight, and the Fourier transform analyzers.
Magnetic Sector Analyzers
Ions generated in the ion source are collimated into a beam through the focusing action of a magnetic field and a slit assembly. After exiting the source, ions are subjected to a magnetic field perpendicular to the direction of the beam. Each ion experiences a force at right angles to both its direction of travel and the direction of the magnetic field, thereby deflecting the beam. The motion of each ion is given by
where m is the mass in atomic mass units, z is the number of electronic charges, H is the magnetic field strength in gauss, r is the radius of the ion trajectory in centimeters, and V is the accelerating voltage. The mass spectrum is scanned by varying the strength of the magnetic field and detecting those ions passing through an exit slit as they come into “focus.” The magnetic sector mass spectrometer affords spatial resolution of ions, giving a unique trajectory at a given field strength for each value of m/z.
Quadrupole Analyzers
The instrument is based on four parallel rods in a square array. The ion beam is focused down the axis of the array and an electrical potential of fixed (DC) and radio frequency (RF) components is applied to diagonally opposed rods. For a given combination of DC and RF components, ions of one specific m/z ratio have a stable path down the axis. All others are deflected to the sides and lost. Mass scanning is achieved by changing the DC and RF components of the voltage, while maintaining a fixed ratio. The quadrupole analyzer is a mass filter because it separates ions on the basis of their m/z ratio.
Ion-trap Analyzer
This quadrupole-type device is composed of a ring electrode placed between two end cap electrodes. Depending upon the commercial version employed, the end caps are either held at ground potential or have an RF voltage applied to them, while an RF voltage is placed on the ring electrode. As a result of these potentials, the hyperbolic surfaces of the three elements form a three-dimensional quadrupole analyzer.
Both ionization and mass analysis take place within the three-dimensional quadrupole field. In the ionization step, the RF voltage on the ring electrode is set low enough so that the ions within the mass range of interest are trapped within the device. Following ionization, mass analysis is accomplished through use of the “mass selective instability” mode of operation. That is, by raising the RF voltage on the ring electrode, ions of successively higher mass are ejected from the ion trap into an electron multiplier detector. In its most common application, the ion-trap analyzer is used in conjunction with a gas chromatograph and covers the mass range of 10 to 560 daltons. However, recent advances in ion-trap technology have extended the workable mass range to many thousands of daltons.
Time-of-flight Analyzers
Ion separation is based on the principle that ions of different masses, possessing equal kinetic energy, have different velocities. If there is a fixed distance for the ions to travel, the time of travel will vary with their mass, the lighter ions traveling faster and reaching the detector in a shorter period of time. The time-of-flight is given by
where tf is the time-of-flight in seconds. Thus, the time-of-flight of the various ions is simply proportional to the square root of the mass-to-charge ratio of the ions. To measure the time-of-flight, ions are introduced into the mass spectrometer in discrete packets so that a starting point for the timing process can be established. Ion packets are generated either through a pulsed ionization process or through a gating system in which ions are produced continuously, but are introduced only at given times into the flight tube.
Fourier Transform Analyzers
In a magnetic field of flux density
B, ions move in circular orbits. The angular frequency,
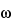
, of the orbital motion is given by
In this type of mass spectrometer, the orbits are varied by subjecting the ions to a resonant alternating electric field. When the frequency of the alternating field matches the orbital frequency, the ions are steadily accelerated to larger and larger orbits in coherent motion, developing a high level of kinetic energy. After the alternating electric field is turned off, the orbiting ions give rise to an alternating image current on the electrodes. A frequency analysis of this signal yields the mass of the ions involved. Thus, the Fourier transform of the time domain transient signal yields the corresponding frequency spectrum from which the mass spectrum is computed. This is a high resolution technique, yielding m/z ratios accurate to about one thousandth of a dalton.
TANDEM MASS SPECTROMETRY
Two mass spectrometers connected in series (MS/MS), tandem mass spectrometry, refers to the use of two or more sequential mass analysis steps. In its simplest form MS/MS (
Figure 2)
Fig. 2. Tandem Mass Spectrometry.
consists of two mass spectrometers linked in such a way that ions preselected by the first mass analyzer (MS1) are chemically or energetically modified and the results analyzed by the second mass analyzer (MS2).
The basic concept of MS/MS involves the ability to determine the mass relationship between a precursor ion in MS1 and a product ion in MS2. Different mass relationships can be probed depending on how MS1 and MS2 are scanned. These include fragmentation of a precursor and measurement of all its fragments (a product scan), selection of multiple precursors and testing for a common fragment (a precursor scan), or scanning to see if a number of precursors all lose the same neutral species (a constant neutral loss scan).
Fragmentation of the precursor ion can be induced by momentum transfer through collision with gas molecules and/or solid surfaces or by electronic excitation using lasers. These techniques are known as collision-induced dissociation, surface-induced dissociation, or laser-induced dissociation, respectively. Allowing the ion to fragment without additional activation is known as metastable decomposition.
There are many applications of MS/MS to pharmaceutical problems. Product scans can be used to obtain qualitative information from precursor ions of drug substances, impurities, and contaminants. This can aid in the identification of unknowns. The method can also be used to determine the amino acid sequence of peptides and protein fragments.
MS/MS has advantages for mixture analysis. Even when the mass spectrometer is coupled to a separation device such as a liquid or gas chromatograph, the resulting signals may be a result of overlapping or unresolved components. MS/MS can be employed to select the precursor ion from one component and obtain structural information without interference from the others.
Selected reaction monitoring is used to reduce the interference encountered during quantitative analysis for low levels of drugs in biological matrices, as in pharmacokinetic studies. If analysis is for a drug specific ion, interfering signals from other compounds in the matrix can mask the desired signal. Interference is reduced if a drug-specific fragment is selected with MS1 and a structure-specific fragment with MS2. The odds of another molecule producing the same mass relationship are diminishingly small.
MS/MS can also be used in metabolism studies to search for molecules with common structural features such as metabolites related to the drug substance. All of the metabolites might contain the same functional group that is lost as a neutral fragment. In this case a contant-neutral-loss scan will show all of these species. For instance, carboxylic acids will all lose neutral carbon dioxide. If the common functionality is lost as an ionic fragment, then a precursor scan will show all of the molecules that produce that fragment ion.
DATA ANALYSIS AND INTERPRETATION
The mass spectral experiment yields information on the molecular weight of ions derived from the sample and the relative abundance of each of these ions. Spectra are often complex, and not all of the ions may be separated by the mass spectrometer. The ability of the instrument to separate ions is called the resolving power, commonly described by the “10% valley” definition. This states that the resolving power is the highest mass number at which two peaks differing by one molecular weight unit and of equal height have a valley between them that is equal to 10% of the peak height. For low, medium, and high resolution mass spectrometers, this value is between 100 and 2000, 2000 and 10,000, and greater than 10,000, respectively.
If one electron is removed or added to a neutral molecule, a molecular ion of essentially the same molecular weight as the parent molecule results. It is often possible to determine the mass of this ion with sufficient precision to enable the empirical formula of the compound to be calculated. Molecular masses may be determined accurately by using high resolution instruments or by peak-matching measurements using reference compounds.
Fragment ions are those produced from the molecular ion by various bond cleavage processes. Numerous papers in the literature relate bond cleavage patterns (fragmentation patterns) to molecular structure.
In addition to measurement of the mass of a molecular ion and its associated fragment ions, mass spectrometers are also used to quantitate compounds with a high degree of selectivity, precision, and accuracy. Compounds are introduced into the mass spectrometer either via direct insertion probe, gas inlet, or, as is more common, via gas or liquid chromatographic interfaces, which provide sample purification. Ionization may be by EI, CI, FAB, thermospray, or electrospray and mass separation by magnetic sector, quadrupole, or quadrupole ion-trap mass spectrometers. Quantitative mass spectrometry involves measuring the abundance of a specific ion, or set of ions, and relating the response to a known standard. External or internal standards may be used, but the latter are preferred for greater accuracy.
For mass spectrometry, internal standards may be either structural or stable isotope analogs. The former have the advantage of lower cost and availability while precision and accuracy are typically achieved by use of a stable isotope (2H, 13C, 15N) labeled analog of the analyte. The only requirements for labeling the analyte are that the ion monitored for the internal standard must retain an isotopic label after ionization and the label must not be exchangeable under the sampling, separation, or ionization conditions. Stable isotope internal standards are often required for acceptable quantitation, particularly with FAB and LC/MS techniques such as thermospray and electrospray.
Relative abundances of the analyte and internal standard ions are typically determined by selected ion monitoring, by which only specific ions due to the analyte and the internal standard are monitored. This technique has the advantage over scanning the full mass range in that more time is spent integrating the ion current at a selected mass-to-charge ratio, thereby increasing sensitivity. Chromatographic peak area or amount of analyte in a sample is calculated from the ratio of analyte to internal standard peak area (or height) and the regression parameters as determined by a calibration curve, using standard techniques.
;)
;)
;)
;)
;)
;)
;)
;)
;)