 727
727 CAPILLARY ELECTROPHORESIS
CAPILLARY ELECTROPHORESIS
Electrophoresis refers to the migration of charged electrical species when dissolved or suspended in an electrolyte through which an electric current is passed. Cations migrate toward the negatively charged electrode (cathode), while anions are attracted toward the positively charged electrode (anode). Neutral particles are not attracted toward either electrode.
The use of capillaries as a migration channel in electrophoresis has enabled analysts to perform electrophoretic separations on an instrumental level comparable to that of high-performance liquid chromatography (HPLC), albeit with some distinct operational differences, advantages, and disadvantages relative to HPLC. This method of analysis is commonly known as capillary electrophoresis (CE). During typical CE operation with an uncoated capillary filled with a buffer, referred to as the “operating buffer,” silanol groups present on the inner wall of the glass capillary release hydrogen ions to the buffer and the wall surface becomes negatively charged, even at a fairly low pH. Cations, or solutes having partial positive charges in the medium, are electrostatically attracted to the negatively charged wall, forming an electrical double layer. The initiation of electrophoresis by applying voltage across the length of the capillary causes the solution portion of the electrical double layer to move toward the cathode end of the capillary, drawing the bulk solution. This movement of the bulk solution under the force of the electrical field is called the electroosmotic flow (EOF). The degree of ionization of the inner-wall capillary silanol groups depends mainly on the pH of the operating buffer and on the modifiers that may have been added to the electrolyte. At low pH, the silanol groups generally have a low ionization and the EOF is low. At higher pH, silanol groups become more ionized and the EOF increases. In some cases organic solvents, such as methanol or acetonitrile, are added to the aqueous buffer to increase the solubility of the solute and other additives or to affect the degree of ionization of the sample. The addition of such organic modifiers generally causes a decrease in the EOF. The detector is located toward the cathode end of the capillary. The EOF is usually greater than the electrophoretic mobility; thus, even anions are swept toward the cathode and the detector. When an uncoated capillary containing pH 7.0 phosphate buffer is used, the usual order of appearance of solutes in an electropherogram is cationic species, neutral solutes, and anionic species.
Currently, there are five major modes of operation of CE: capillary zone electrophoresis (CZE), also referred to as free solution or free flow capillary electrophoresis; micellar electrokinetic chromatography (MEKC); capillary gel electrophoresis (CGE); capillary isoelectric focusing (CIEF); and capillary isotachophoresis (CITP).
In CZE, separations are controlled by differences in the relative electrophoretic mobilities of the individual components in the sample or test solution. The mobility differences are functions of analyte charge and size under specific method conditions. They are optimized by appropriate control of the composition of the buffer, its pH, and its ionic strength.
In MEKC, ionic surfactants are added to the operating buffer at a concentration above their critical micelle concentration. The micelles provide a pseudostationary phase with which analytes can partition. This technique is useful for the separation of neutral and ionic species.
CGE, which is analogous to gel filtration, uses gel-filled capillaries to separate molecules on the basis of relative differences in their respective molecular weight or molecular size. It was first used for the separation of proteins, peptides, and oligomers. Gels may have the advantage of decreasing the EOF and also significantly reducing protein adsorption onto the inner wall of the capillary, which can significantly reduce analyte peak tailing effects.
In CIEF, substances are separated on the basis of their relative differences in isoelectric points. This is accomplished by achieving steady-state sample zones within a buffer pH gradient, where the pH is low at the anode and high at the cathode. The gradient is established by applying a voltage across a capillary filled with a mixture of carrier components consisting of amphoteric substances having different pI values.
CITP employs two buffers that enclose the analyte zones between them. Either anions or cations can be analyzed in sharply separated zones. In addition, the analyte concentrations are the same in each zone; thus, the length of each zone is proportional to the amount of the particular analyte.
The most commonly utilized capillary electrophoresis techniques are CZE and MEKC. These are briefly discussed in the following sections. Pertinent general principles and theory, instrumental considerations, analysis, and operational considerations and parameters are discussed as well.
PRINCIPLES OF CAPILLARY ZONE ELECTROPHORESIS
CZE makes use of the principles of electrophoresis and electroosmosis to achieve separation of charged species.
(1) The electrophoretic mobility of an ion, µEP, is described by the equation:
µ
EP =
q / (6
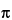
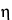 r
r),
in which
q is the charge of the ion,
is the solution viscosity, and
r is the radius of the hydrated ion. This relationship infers that small, highly charged analytes have high mobilities and large, slightly charged analytes have low mobilities.
(2) The velocity of migration,
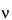 EP
EP, in cm per second, is represented by the equation:
EP = µ
EP(
V /
L),
in which µEP is the electrophoretic mobility; V is the applied voltage; and L, in cm, is the total capillary length.
(3) The velocity of the EOF,
EO, in cm per second, is described by the equation:
EO = µ
EO(
V /
L),
in which µEO is the EOF mobility (the coefficient of electroosmotic flow), and the other terms are as defined above.
(4) The time, t, in seconds, necessary for a solute to migrate the entire effective length of the capillary (from the inlet to the detector), l, is represented by the relationship:
t = l / E(µEP + µEO) = lL / V(µEP + µEO),
in which E is the strength of the applied electrical field, and the other terms are as defined above.
(5) Efficiency of an electrophoretic system can be related to mobility and EOF and expressed in terms of the number of theoretical plates, N, by the equation:
N = (µEP + µEO)V / 2D,
in which D is the diffusion coefficient of the solute, and the other terms are as defined above.
(6) The resolution, R, of two consecutively eluting solutes can be defined by the equation:
where µEP1 and µEP2 are the mobilities of the two solutes,
is their average, and the other terms are as defined above.
PRINCIPLES OF MICELLAR ELECTROKINETIC CHROMATOGRAPHY
In MEKC, the supporting electrolyte medium contains a surfactant at a concentration above its critical micelle concentration (CMC). In this aqueous medium, the surfactant self-aggregates and forms micelles whose hydrophilic head groups form an outer shell and whose hydrophobic tail groups form a nonpolar core into which the solutes can partition. Generally, the micelles are anionic on their surface, and, under the applied voltage, they migrate in the opposite direction to the EOF. This type of partitioning is analogous to that in solvent extraction or reverse-phase HPLC. The differential partitioning of neutral molecules between the buffered aqueous mobile phase and the micellar pseudostationary phase is the sole basis for separation. The buffer and micelles form a two-phase system, and the analyte can partition between these two phases.
A micellar system suitable for MEKC meets the following criteria: the surfactant is highly soluble in the buffer, and the micellar solution is homogeneous and transparent when UV detection is employed. The most common surfactant for MEKC is sodium dodecyl sulfate (anionic surfactant). Others include cetyltrimethylammonium bromide (cationic surfactant) and bile salts (chiral surfactant). The selectivity of an MEKC system is mainly dependent on the nature of the surfactant. Organic solvents are often added to the MEKC buffer to adjust the capacity factors, just as in reverse-phase HPLC separations. MEKC may be used for the separation of enantiomers. For such separations, a chiral additive is added to the buffer or a chiral surfactant, such as a bile salt, is used.
A general knowledge of conventional column chromatographic principles aids in understanding MEKC principles. However, in MEKC the micelles are not truly stationary; therefore, the column chromatographic theory needs to be modified. The major modification introduced to MEKC principles is the finite nature of the separation window for neutral molecules.
(7) The migration time, tR, for a neutral species is expressed with the following equation:
tR = (1 + k¢)t0 / [1 + (t0 / tMC)],
in which t0 is the time required for an unretained substance to travel the effective length of the capillary; tMC is the time required for a micelle to traverse the capillary; k¢ is the capacity factor; and tR is always between t0 and tMC.
(8) The capacity factor, k¢, for a neutral species, is calculated by the equation:
k¢ = (
tR /
t0 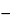
1) / (1
tR /
tMC),
in which the terms are as defined above.
(9) For practical purposes, k¢ is calculated by the equation:
k¢ =
tR /
t0 1,
in which tR is the time measured from the point of voltage application (or injection) to the peak maximum; and t0 is measured from the point of voltage application (or injection) to the leading edge of the solvent front or of an unretained substance. In contrast with CZE, k¢ in MEKC is significant and is a characteristic of a given solute in a given MEKC system. Further discussion of k¢ appears later in the System Suitability section under Operational Parameters.
(10) The resolution, RS, for neutral species is calculated by the equation:
in which
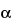
is the selectivity, defined as the ratio of
k¢2 to
k¢1, of the operating conditions for separating two solutes. If the two solutes elute close together (
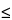
1.1), either
k¢ may be used. The equation shows that, just as with conventional chromatography, resolution in MEKC can be improved through controlling efficiency, selectivity, retention, and the chemical nature of the resolving surfactant-medium system. The last term of the equation is due to the limited elution range. Although MEKC is particularly useful in the separation of neutral species, this technique may also be used for the separation of charged solutes. The latter procedure involves a combination of chromatographic and electrophoretic separation mechanisms. The additional interaction between charged solutes and micelle can be used to optimize a separation. Ion-pairs may form if the charges borne on the surfactant and solute are opposite; otherwise, surfactant and solute repel each other. These differences can significantly influence the separation of charged molecules.
INSTRUMENTAL CONSIDERATIONS
A typical CE system (see
Figure 1) contains a fused-silica capillary having an internal diameter of 50 to 100 µm and a length of 20 to 100 cm. The ends of the capillary are placed in separate electrolyte reservoirs. The direct-current power supply is capable of furnishing high voltages, typically ranging from 0 to 30 kV. A detector and autosampler with some form of data-recording device complete the system. An automatic buffer replenishment system and a computer-based control and data acquisition system may also be found on the standard commercial systems. Temperature controls for both the capillary and the autosampler are also available on commercial instruments.
The primary considerations of instrumentation include capillary type and configuration, modes of sampling, power supply and detector modes.
Fig. 1. Typical CE Instrument Configuration.
Capillary Type and Configuration
Capillaries used in CZE are usually made of fused silica and with no internal coating. Some instruments are configured with a “free-swinging” style of capillary; that is, the capillary is not encased within an enclosure. In most commercial instruments, the capillary is housed in a cartridge. Both configurations offer specific advantages and disadvantages. The ability of the instrument to accommodate different types of capillaries and capillaries of various diameters and lengths is an important consideration. Capillaries with a variety of internal coatings are also available; therefore, the ability of the instrument to accommodate different capillaries is important. Internal capillary coatings may be employed to alter the magnitude or direction of EOF or to reduce sample absorption. If an internally coated capillary is to be used, then sufficient details and the indication of the supplier must be included in the method. Capillaries from an alternate supplier can be used if it is demonstrated that they are suitable.
Sample Introduction and Injector Technology
Modes of sample introduction onto the capillary include electromigration (electrokinetic mode) and negative- and positive-pressure injection (hydrostatic mode).
For injection via electromigration, the sample solution is electrophoresed into the capillary by inserting the capillary and electrode into the sample vials and applying a brief, high voltage. The sample enters the capillary by a combination of electrophoresis and EOF. Therefore, analytes with different mobilities are loaded into the capillary to different extents. The conductivities of the sample and standard solutes also affect the EOF and the volume injected.
Negative-pressure injectors place negative pressure at the detector end of the capillary and draw the sample solution into the injection end of the capillary. Positive-pressure injectors pressurize the sample vial, forcing the sample into the capillary. Pressure injection loads all sample components into the capillary to the same extent, and it is generally the most reproducible and the most frequently applied injection mode. The sample volume injected depends on the capillary length and internal diameter and the voltage or pressure applied. The typical sample volumes injected into the capillary are between 1 and 20 nL.
Each injection method offers specific advantages and disadvantages, depending on the sample composition, the separation mode, and the application of the method. None of the above injection modes is as reproducible as commercially available HPLC injectors. Based on the circumstances, it may be necessary to use internal standards for specific methods where high injection precision is required.
Power Supply
Most commercially available CE units have direct-current power supplies that are capable of furnishing power on a ramp-up or step-function mode to achieve and maintain the desired operational voltage in a smooth manner. This will help to ensure a relatively smooth baseline.
Another essential feature of the power supply is its utility in introducing a sample at the cathodic or the anodic end of the capillary. Because it is impractical to relocate the on-line detector from one end of the instrument to the other, it is beneficial to be able to specify whether the sample injection end is at the cathode or the anode.
Detector Modes
CE systems generally offer UV-visible absorbance and laser-induced fluorescence (LIF) detectors. Scanning UV detectors or photodiode-array detectors are also available for many commercial CE instruments.
The coupling of CE to a mass spectrometer offers the possibility of obtaining structural information in conjunction with electrophoretic migration data.
Fluorescence detection offers an enhanced sensitivity for samples containing only very small amounts of UV-active analytes. Application of fluorescent tags to non-UV-absorbing compounds can be useful. Alternately, non-UV-absorbing or nonfluorescent analytes can be detected indirectly by adding a chromophore or a fluorophore, respectively, to the buffer: the non-absorbing species are detected through the absence of expected signal from the absorbing species. Conductivity and pulsed amperometric detectors can also be used but are not generally available on commercial CE instruments.
ANALYTICAL CONSIDERATIONS
Several parameters, namely, capillary dimensions, voltage, ionic strength, and pH, are optimized to give adequate resolution and separation. Care should be taken to avoid changes in temperature that will affect the viscosity of the buffer and, in turn, influence both the EOF and the solute mobilities.
Capillary Dimensions—
Variation of the capillary diameter and length can affect the electrophoretic resolution. Increasing the capillary length results in longer migration times, usually increasing resolution and generating a lower current. Increasing the capillary diameter will usually increase current and associated internal temperature gradients that decrease resolution. Conversely, a reduction in capillary diameter will result in lower heat and better resolution. However, larger capillary diameters have advantages of better mass loading and improved signal-to-noise ratio.
Voltage Effects—
When higher voltages are applied, additional internal heating of the operating buffer occurs because of the current flow through the buffer. This heating effect, known as Joule heating, must be controlled because resistance, dielectric constant, and viscosity are temperature-dependent and alter the velocity of the EOF and solute mobilities.
In general, increasing the voltage will result in increased efficiency and resolution (up to the point where Joule heat cannot be adequately dissipated). Maximum resolution is obtained by maintaining the voltage below the level at which Joule heating and diffusion become limiting factors.
Ionic Strength Effects—
Control of ionic strength and its manipulation allow adjustment of resolution, efficiency, and sensitivity. Increasing ionic strength will generally improve resolution, peak efficiency, and peak shape. Sensitivity may be improved because better focusing is achieved. However, because the current generated is directly proportional to the buffer concentration, more heat is produced when ionic strength of the buffer is increased, hence limiting the ionic strengths that can be utilized.
pH Effects—
Resolution, selectivity, and peak shape can be dramatically altered by changes in pH as this parameter affects the extent of solute ionization and the level of EOF. The EOF is high at high pH and low at low pH in uncoated fused-silica capillaries.
OPERATIONAL PARAMETERS
The major steps in operating a CE system are system setup, capillary rinsing procedure, running a sample, system suitability testing, sample analysis, data handling, and system shutdown.
System Setup—
An appropriate capillary of specific length, inner diameter, and coating is selected, with considerations made for separation and resolution, ionic strength of buffer, and pH effects. A buffer of appropriate composition, ionic strength, and pH is prepared, degassed, if necessary, and passed through an appropriate filter. All solvents, including water, are HPLC or CE grade.
Capillary Rinsing Procedure—
Improved consistency of migration times and resolution may generally be obtained if a defined rinsing procedure is followed. Capillary conditioning and rinsing procedures are very specific to the analyte, matrix, and method. Therefore, these procedures are developed as part of the method and are specified in the individual monograph. Rinsing may involve the use of solutions such as 0.1 M phosphoric acid, water, and 0.1 M sodium hydroxide. Before beginning analysis of the test specimen, the capillary may be rinsed with five column volumes of the operating buffer that is to be used for the test. When changing buffer composition, it is advisable to rinse the capillary with five column volumes of each new buffer to allow the capillary to be cleansed of the previous buffer. Use of a new uncoated fused-silica capillary usually requires a regeneration procedure to activate the surface silanol groups. This procedure may include an extended rinse with a sodium hydroxide solution. Coated capillaries are rinsed according to the manufacturer's guidelines because inappropriate rinsing can remove or damage the coating. Columns may be dedicated to particular methods or buffer types to prevent cross-contamination.
Running a Sample—
An appropriate capillary, electrolyte, and injection procedure are selected to achieve adequate resolution, sensitivity, and separation, with well-shaped and well-defined peaks. The required injection precision for a specific method may require use of an internal standard. The internal standard is selected with consideration of its ability to adequately separate from the analyte. The performance of the system may be improved by rinsing the capillary between injections and supplying fresh buffer to the source and destination vials used during voltage application, namely, vials 2 and 4 in
Figure 1. Replicate injections from the same sample vial may be performed provided that no cross-contamination occurs. If cross-contamination occurs, the capillary tip may be rinsed by briefly inserting it into a vial containing the buffer prior to inserting the capillary into the electrolyte or sample vial.
The operational parameters are specified in each individual monograph so as to minimize voltage effects, ionic strength effects, and pH effects. The instrument is set up to run with the appropriate capillary configuration and injection conditions, within the established linear dynamic range of the detector; and acceptable migration precision is ensured by appropriate choice of sample diluent, separation electrolyte, electrolyte additives, and capillary pretreatment conditions. Exercise caution to avoid overloading the capillary with sample, as this decreases efficiency and reproducibility.
System Suitability—
Parameters measured may include injector reproducibility, system selectivity, system efficiency, and tailing. Resolution between the analytes and other compounds may be determined by using test mixture standards.
Parameters typically used to determine system suitability include relative standard deviation (RSD), capacity factor (k¢), the number of theoretical plates (N), sensitivity (limit of detection or quantitation), number of theoretical plates per meter (TPM), tailing factor (T), and resolution (R).
The peak shape is closely examined; ideally, the peak is symmetrical, with no shoulders and no excessive tailing. If these conditions are not met, corrective actions are taken before proceeding with the analysis. Peak integration is also closely examined to ensure that the peak response is correctly quantitated.
Replicate injections of a Standard preparation of known concentration can be used to determine the reproducibility of the CE system. Data from five or more replicate injections are used to calculate RSD. Unless otherwise specified in the individual monograph, the relative standard deviation for replicate injections is not more than 3.0%. Minimum injection precision values may be specified in specific CE methods, especially when determining trace-level components. Calculation of electrophoretic parameters in MEKC, as in other forms of CE, may involve a combination of chromatographic and electrophoretic relationships. Hence, capacity factor, k¢, for neutral analyte migration in MEKC can be calculated by the equation:
k¢ = tR – t0(1 – tR / tMC),
in which tR, t0, and tMC are the migration times of the analyte, the bulk solution (EOF), and the micelle, respectively.
The number of theoretical plates, N, is a measure of the efficiency of the system and is calculated by the equation:
N = 16(tR / W)2 or N = 5.54(tR / W1/ 2)2,
in which W is the analyte peak width at baseline, W1/2 is the analyte peak width at half-height, and tR is the analyte migration time.
The number of theoretical plates per meter, TPM, is a measure of the efficiency of the capillary as a function of peak width at baseline and can be calculated by the equation:
TPM = 1600(tR / W)2 / L,
in which L, in cm, is the total capillary length; and the other terms are as defined above. The tailing factor, T, of the analyte peak is a measure of peak symmetry, and it represents the degree of deviation of the symmetry of the peak from an ideally symmetrical Gaussian peak. This factor can be calculated by the equation:
T = W0.05 / 2f,
in which W0.05 is the length of a line constructed parallel to the peak base from the leading edge to the tailing edge of the peak at 5% of peak height; and f is the distance along the same line from the leading edge of the peak, appearing to the left of the peak maximum in the electropherogram, to the intercept of a perpendicular line dropped from the peak maximum to the base. A ratio of 1.0 indicates a perfectly symmetrical peak. If electrodispersive effects occur, they can generate highly asymmetrical peaks. This can occur when high sample concentrations are used, such as those for testing of impurities. Use of highly asymmetrical peaks is acceptable provided that they are reproducible and that they do not compromise separation selectivity.
The resolution factor, R, is a measure of the ability of the capillary system to separate consecutively migrating analytes. Resolution is determined for all sample analytes of interest, with the pH of the buffer adjusted as necessary to meet system suitability requirements. It can be calculated by the equation:
R = 2(
t2 t1) / (
W1 +
W2),
in which t2 and t1 are the migration times, measured at peak maxima, for the slower migrating peak and the faster migrating peak, respectively; and W2 and W1 are the corresponding widths of these two peaks measured at their bases.
Sample Analysis—
Once the suitability of the CE system has been established, aliquots of both the Standard preparation and the test preparation are injected. Standards are injected before or after the samples and intermittently throughout the run.
Data Handling—
Time-normalized peak areas are often used in quantitative calculations. These are determined by dividing the observed integrated peak area by the migration time of the analyte. This compensates for the fact that in CE, unlike HPLC, each analyte travels through the detector at a different velocity. Unless this normalization is performed, slowly moving (later-migrating) analytes will have disproportionately large peak areas compared with those for early migrating components.
System Shutdown—
After analysis, the capillary is rinsed according to the directions specified in each monograph or as recommended by the manufacturer. For example, the capillary might be rinsed with distilled water to remove buffer components and then filled with air or nitrogen by performing a rinse from an empty vial. Naturally, the destination and source vials, namely, vials 4 and 2 in
Figure 1, are emptied of buffer and rinsed thoroughly with deionized water.
;)
;)
;)
;)