Direct Titrations—
Direct titration is the treatment of a soluble substance, contained in solution in a suitable vessel (the titrate), with an appropriate standardized solution (the titrant), the endpoint being determined instrumentally or visually with the aid of a suitable indicator.
The titrant is added from a suitable buret and is so chosen, with respect to its strength (normality), that the volume added is between 30% and 100% of the rated capacity of the buret. [NOTE—Where less than 10 mL of titrant is required, a suitable microburet is to be used.] The endpoint is approached directly but cautiously, and finally the titrant is added dropwise from the buret in order that the final drop added will not overrun the endpoint. The quantity of the substance being titrated may be calculated from the volume and the normality or molarity factor of the titrant and the equivalence factor for the substance given in the individual monograph.
Oxidation-Reduction (Redox) Titrations—
Determinations may often be carried out conveniently by the use of a reagent that brings about oxidation or reduction of the analyte. Many redox titration curves are not symmetric about the equivalence point, and thus graphical determination of the endpoint is not possible; but indicators are available for many determinations, and a redox reagent can often serve as its own indicator. As in any type of titration, the ideal indicator changes color at an endpoint that is as close as possible to the equivalence point. Accordingly, when the titrant serves as its own indicator, the difference between the endpoint and the equivalence point is determined only by the analyst's ability to detect the color change. A common example is the use of permanganate ion as an oxidizing titrant since a slight excess can easily be detected by its pink color. Other titrants that may serve as their own indicators are iodine, cerium (IV) salts, and potassium dichromate. In most cases, however, the use of an appropriate redox indicator will yield a much sharper endpoint.
It may be necessary to adjust the oxidation state of the analyte prior to titration through use of an appropriate oxidizing or reducing agent; the excess reagent must then be removed, e.g., through precipitation. This is nearly always the practice in the determination of oxidizing agents since most volumetric solutions of reducing agents are slowly oxidized by atmospheric oxygen.
Titrations in Nonaqueous Solvents—
Acids and bases have long been defined as substances that furnish, when dissolved in water, hydrogen and hydroxyl ions, respectively. This definition, introduced by Arrhenius, fails to recognize the fact that properties characteristic of acids or bases may be developed also in other solvents. A more generalized definition is that of Brönsted, who defined an acid as a substance that furnishes protons, and a base as a substance that combines with protons. Even broader is the definition of Lewis, who defined an acid as any material that will accept an electron pair, a base as any material that will donate an electron pair, and neutralization as the formation of a coordination bond between an acid and a base.
The apparent strength of an acid or a base is determined by the extent of its reaction with a solvent. In water solution all strong acids appear equally strong because they react with the solvent to undergo almost complete conversion to oxonium ion and the acid anion (leveling effect). In a weakly protophilic solvent such as acetic acid the extent of formation of the acetate acidium ion shows that the order of decreasing strength for acids is perchloric, hydrobromic, sulfuric, hydrochloric, and nitric (differentiating effect).
Acetic acid reacts incompletely with water to form oxonium ion and is, therefore, a weak acid. In contrast, it dissolves in a base such as ethylenediamine, and reacts so completely with the solvent that it behaves as a strong acid. The same holds for perchloric acid.
This leveling effect is observed also for bases. In sulfuric acid almost all bases appear to be of the same strength. As the acid properties of the solvent decrease in the series sulfuric acid, acetic acid, phenol, water, pyridine, and butylamine, the bases become progressively weaker until all but the strongest have lost their basic properties. In order of decreasing strength, the strong bases are sodium 2-aminoethoxide, potassium methoxide, sodium methoxide, and lithium methoxide.
Many water-insoluble compounds acquire enhanced acidic or basic properties when dissolved in organic solvents. Thus the choice of the appropriate solvent permits the determination of a variety of such materials by nonaqueous titration. Furthermore, depending upon which part of a compound is the physiologically active moiety, it is often possible to titrate that part by proper selection of solvent and titrant. Pure compounds can be titrated directly, but it is often necessary to isolate the active ingredient in pharmaceutical preparations from interfering excipients and carriers.
The types of compounds that may be titrated as acids include acid halides, acid anhydrides, carboxylic acids, amino acids, enols such as barbiturates and xanthines, imides, phenols, pyrroles, and sulfonamides. The types of compounds that may be titrated as bases include amines, nitrogen-containing heterocyclic compounds, oxazolines, quaternary ammonium compounds, alkali salts of organic acids, alkali salts of weak inorganic acids, and some salts of amines. Many salts of halogen acids may be titrated in acetic acid or acetic anhydride after the addition of mercuric acetate, which removes halide ion as the unionized mercuric halide complex and introduces the acetate ion.
For the titration of a basic compound, a volumetric solution of perchloric acid in glacial acetic acid is preferred, although perchloric acid in dioxane is used in special cases. The calomel-glass electrode system is useful in this case. In acetic acid solvent, this electrode system functions as predicted by theory.
For the titration of an acidic compound, two classes of titrant are available: the alkali metal alkoxides and the tetraalkylammonium hydroxides. A volumetric solution of sodium methoxide in a mixture of methanol and toluene is used frequently, although lithium methoxide in methanol-benzene solvent is used for those compounds yielding a gelatinous precipitate on titration with sodium methoxide.
The alkali error limits the use of the glass electrode as an indicating electrode in conjunction with alkali metal alkoxide titrants, particularly in basic solvents. Thus, the antimony-indicating electrode, though somewhat erratic, is used in such titrations. The use of quaternary ammonium hydroxide compounds, e.g., tetra-n-butylammonium hydroxide and trimethylhexadecylammonium hydroxide (in benzene-methanol or isopropyl alcohol), has two advantages over the other titrants in that (a) the tetraalkylammonium salt of the titrated acid is soluble in the titration medium, and (b) the convenient and well-behaved calomel-glass electrode pair may be used to conduct potentiometric titrations.
Because of interference by carbon dioxide, solvents for acidic compounds need to be protected from excessive exposure to the atmosphere by a suitable cover or by an inert atmosphere during the titration. Absorption of carbon dioxide may be determined by performing a blank titration. The blank should not exceed 0.01 mL of 0.1 N sodium methoxide VS per mL of solvent.
The endpoint may be determined visually by color change, or potentiometrically, as indicated in the individual monograph. If the calomel reference electrode is used, it is advantageous to replace the aqueous potassium chloride salt bridge with 0.1 N lithium perchlorate in glacial acetic acid for titrations in acidic solvents or potassium chloride in methanol for titrations in basic solvents.
Where these or other mixtures are specified in individual monographs, the calomel reference electrode is modified by first removing the aqueous potassium chloride solution and residual potassium chloride, if any, by rinsing with water, then eliminating residual water by rinsing with the required nonaqueous solvent, and finally filling the electrode with the designated nonaqueous mixture.
In nearly all cases, except those where silver ion might interfere, a silver-silver chloride reference electrode may be substituted for the calomel electrode. The silver-silver chloride electrode is more rugged, and its use helps to eliminate toxic mercury salts from the laboratory. Generally, a salt bridge may be used to circumvent interference by silver ion.
The more useful systems for titration in nonaqueous solvents are listed in
Table 1.
Table 1. Systems for Nonaqueous Titrations
Indicator and Potentiometric Endpoint Detection—
The simplest and most convenient method by which the equivalence point, i.e., the point at which the stoichiometric analytical reaction is complete, may be determined is with the use of indicators. These chemical substances, usually colored, respond to changes in solution conditions before and after the equivalence point by exhibiting color changes that may be taken visually as the endpoint, a reliable estimate of the equivalence point.
A useful method of endpoint determination results from the use of electrochemical measurements. If an indicator electrode, sensitive to the concentration of the species undergoing titrimetric reaction, and a reference electrode, whose potential is insensitive to any dissolved species, are immersed in the titrate to form a galvanic cell, the potential difference between the electrodes may be sensed by a pH meter and used to follow the course of the reaction. Where such a series of measurements is plotted correctly (i.e., for an acid-base titration, pH versus mL of titrant added; for a precipitimetric, complexometric, or oxidation-reduction titration, mV versus mL of titrant added), a sigmoid curve results with a rapidly changing portion (the “break”) in the vicinity of the equivalence point. The midpoint of this linear vertical portion or the inflection point may be taken as the endpoint. The equivalence point may also be determined mathematically without plotting a curve. However, it should be noted that in asymmetrical reactions, which are reactions in which the number of anions reacting is not the same as the number of cations reacting, the endpoint as defined by the inflection of the titration curve does not occur exactly at the stoichiometric equivalence point. Thus, potentiometric endpoint detection by this method is not suitable in the case of asymmetric reactions, examples of which are the precipitation reaction,
2Ag+ + CrO4–2
and the oxidation-reduction reaction,
5Fe+2 + MnO4–.
All acid-base reactions, however, are symmetrical. Thus, potentiometric endpoint detection may be employed in acid-base titrations and in other titrations involving symmetrical reversible reactions where an indicator is specified, unless otherwise directed in the individual monograph.
Two types of automatic electrometric titrators are available. The first is one that carries out titrant addition automatically and records the electrode potential differences during the course of titration as the expected sigmoid curve. In the second type, titrant addition is performed automatically until a preset potential or pH, representing the endpoint, is reached, at which point the titrant addition ceases.
Several acceptable electrode systems for potentiometric titrations are summarized in
Table 2.
Table 2. Potentiometric Titration Electrode Systems
| Titration |
Indicating Electrode |
Equation1 |
Reference Electrode |
Applicability2 |
| Acid-base |
Glass |
E = k + 0.0591 pH |
Calomel or silver–silver
Chloride |
Titration of acids and bases |
| Precipitimetric (silver) |
Silver |
E = E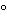 + 0.0591 log [Ag +] + 0.0591 log [Ag +] |
Calomel (with
 potassium nitrate potassium nitrate
salt bridge) |
Titration with or of silver
involving halides
or thiocyanate |
| Complexometric |
Mercury–mercury(II) |
E = E + 0.0296(log k¢
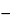 pM) pM) |
Calomel |
Titration of various metals
(M), e.g., Mg+2, Ca+2
Al+3, Bi+3, with EDTA |
| Oxidation–reduction |
Platinum |
E = E + (0.0591/n)
×log [ox]/[red] |
Calomel or silver–silver
chloride |
Titrations with arsenite,
bromine, cerate,
dichromate,
exacyonoferrate(III),
iodate, nitrite,
permanganate,
thiosulfate |
1
Appropriate form of Nernst equation describing the indicating electrode system: k = glass electrode constant; k¢ = constant derived from Hg–Hg(II)–EDTA equilibrium; M = any metal undergoing EDTA titration; [ox] and [red] from the equation, ox + n e 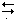 red.
2
Listing is repesentative but not exhaustive.
|