Capillary electrophoresis is a physical method of analysis based on the migration, inside a capillary, of charged analytes dissolved in an electrolyte solution, under the influence of a direct-current electric field. In this section we are describing four capillary electrophoresis methods, Free Solution Capillary Electrophoresis, Capillary Gel Electrophoresis, Capillary Isoelectric Focusing, and Micelle Electrokinetic Chromatography.
General Principle
The migration velocity of the analyte under an electric field of intensity, E, is determined by the electrophoretic mobility of the analyte and the electroosmotic mobility of the buffer inside the capillary. The electrophoretic mobility of a solute (µep) depends on the characteristics of the solute (electrical charge, molecular size, and shape) and the characteristics of the buffer in which the migration takes place (type and ionic strength of the electrolyte, pH, viscosity, and additives). The electrophoretic velocity (Vep) of a solute, assuming a spherical shape, is as follows:
in which
q is the effective charge of the particle;
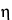
is the viscosity of the buffer;
r is the size of the solute ion;
V is the applied voltage; and
L is the total length of the capillary.
When an electric field is applied through the capillary filled with buffer, a flow of solvent is generated inside the capillary called electroosmotic flow. Its velocity depends on the electroosmotic mobility (µeo) which in turn depends on the charge density on the capillary internal wall and the buffer characteristics. The electroosmotic velocity (Veo) is as follows:
in which
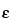
is the dielectric constant of the buffer;
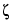
is the zeta potential of the capillary surface; and the other terms are as defined above.
The electrophoretic and electroosmotic mobilities of the analyte may act in the same direction or in opposite directions, depending on the charge (positive or negative) of the solute, and the velocity of the solute (v) is as follows:
V = Vep ± Veo.
The sum or the difference between the two velocities (Vep and Veo) is used depending on whether the mobilities act in the same or opposite directions. Under conditions with a fast Veo, with respect to the Vep of the solutes, both negative and positive charged analytes can be separated in the same run. The time (t) taken by the solute to migrate the distance (l) from the injection end of the capillary to the detection point (capillary effective length) is as follows:
in which the other terms are as defined above.
In general, the fused-silica capillaries used in electrophoresis bear negative charges on the inner wall, producing electroosmotic flow towards the cathode. The electroosmotic flow has to remain constant from run to run to obtain good reproducibility in the migration velocity of the solutes. For some applications, it might be necessary to reduce or suppress the electroosmotic flow by modifying the inner wall of the capillary or by changing the pH of the buffer solution.
When the sample is introduced in the capillary, each analyte ion of the sample migrates within the background electrolyte as an independent zone according to its electrophoretic mobility. The spreading of each solute band (zone dispersion) results from a different phenomena. Under ideal conditions, the sole contribution to the solute-zone broadening is molecular diffusion of the solute along the capillary (longitudinal diffusion). In this case, the efficiency of the zone is expressed as the number of theoretical plates (N), as follows:
in which D is the molecular diffusion of the solute in the buffer; and the other terms are as defined above.
From a practical point of view, other phenomena such as heat dissipation, sample adsorption onto the capillary wall, mismatched conductivity between sample and buffer, length of the injection plug, detector cell size, and unleveled buffer reservoirs can also significantly contribute to band dispersion. Separation between two bands (expressed by the resolution, RS) can be achieved by modification of the electrophoretic mobility of the analytes, by the electroosmotic mobility induced by capillary, and by increasing the efficiency for the band of each analyte as follows:
in which µepa and µepb are the electrophoretic mobilities of the two compounds to be separated; bar(µ)ep is the average electrophoretic mobility of the two solutes calculated as:
bar(µ)ep = ½ (µepb + µepa),
and the other terms are as defined above.
Apparatus
An apparatus for capillary electrophoresis is composed of a high voltage controllable power supply; two buffer reservoirs held at the same level and containing specified anodic and cathodic solutions; two electrodes assemblies (cathode and anode) immersed in the buffer reservoirs and connected to the power supply; a separation capillary usually made of fused-silica, with sometimes an optical viewing window aligned with detector, depending on the detector, with the ends of the capillary placed in the buffer reservoirs and the capillary being filled with a solution specified in a given monograph; a suitable injection system; a detector capable of monitoring the amount of substance of interest passing through a segment of the separation capillary at a given time, generally based on absorption spectrophotometry (UV and visible), fluorimetry, conductimetric, amperometric, or mass spectrometric detection, depending on the specific applications, or even indirect detection to detect non-UV-absorbing and nonfluorescent compounds; and a thermostatic system capable of maintaining the temperature inside the capillary.
The method of injection of samples and its automation is critical for precise quantitative analysis. Methods of injection include gravity, pressure or vacuum, or electrokinetic injection. The amount of each sample component introduced electrokinetically depends on its electrophoretic mobility, thus possibly biasing the results.
It is expected that the capillary, the buffer solutions, the preconditioning method, the sample solution, and the migration conditions will be specified in the individual monograph. The electrolytic solution employed may be filtered to remove particles and degassed to avoid bubble formation that could interfere with the detection system. To achieve reproducible migration time of the solutes, if would be necessary to develop, for each analytical method, a rigorous rinsing routine after each injection.
Free Solution Capillary Electrophoresis
In free solution capillary electrophoresis, analytes are separated in a capillary containing only buffer without any anticonvective medium. In this technique, separation takes place because the different components of the sample migrate as discrete bands with different velocities. The velocity of each band depends on the electrophoretic mobility of the solute and the electroosmotic flow on the capillary. Coated capillaries, with reduced electroosmotic flow, can be used to increase the separation capacity of those substances absorbing on fused silica surfaces.
This mode of capillary electrophoresis is appropriate for the analysis of small (MW < 2000) and large molecules (2000 < MW < 100,000). Due to the high efficiency achieved, molecules having only minute differences in their charge-to-mass ratio can be separated. This method also allows the separation of chiral compounds by adding chiral selectors to the separation buffer. The optimization of the separations requires consideration of a number of instrumental and electrolytic solution parameters.
INSTRUMENTAL PARAMETERS
Voltage—
The separation time is universally proportional to applied voltage. However, an increase in the voltage used can cause excessive heat production, giving rise to temperature and viscosity gradients in the buffer inside the capillary, which causes band broadening and decreases resolution.
Temperature—
The main effect of temperature is observed on buffer viscosity and electrical conductivity, thus affecting migration velocity. In some cases, an increase in capillary temperature can cause a conformational change of some proteins, modifying their migration time and the efficiency of the separation.
Capillary—
The length and internal diameter of the capillary affects the analysis time, the efficiency of separations, and the load capacity. Increasing both effective length and total length can decrease the electric fields, at a constant voltage, which will increase migration time. For a given buffer and electric field, heat dissipation (thus sample band broadening) depends on the internal diameter of the capillary. The latter also affects the detection limit, depending on the sample volume injected into the capillary and the detection system used.
The adsorption of sample components on the capillary wall limits efficiency; therefore, methods to avoid these interactions should be considered in the development of a separation method. This is critical in samples containing proteins. Strategies have been devised to avoid adsorption of proteins on the capillary wall. These strategies include both the use of extreme pH and the absorption of positively charged buffer additives that only require modification of the buffer composition. Other strategies include the coating of the internal wall of the capillary with a polymer covalently bonded to the silica that prevents interaction between the proteins and the negatively charged silica surface. Capillaries with coatings consisting of neutral-hydrophilic, cationic, and anionic polymers are commercially available.
ELECTROLYTIC SOLUTION PARAMETERS
Buffer Type and Concentrations—
Suitable buffers for capillary electrophoresis have an appropriate buffer capacity in the pH range of choice and low mobility to minimize current generation.
To minimize peak shape distortion, it is important to match buffer–ion mobility to solute mobility, whenever possible. The type of sample solvent used is important to achieve on-column sample focusing which increases separation efficiency and improves detection. Also, an increase in buffer concentration at a given pH will decrease electroosmotic flow and solute velocity.
Buffer pH—
The pH of the buffer can affect separation by modifying the charge of the analyte or other additives and by changing the electroosmotic flow. For protein and peptide separation, a change in the pH of the buffer from above the isoelectric point to below the isoelectric point changes the net charge of the solute from negative to positive. An increase in the buffer pH generally increases the electroosmotic flow.
Organic Solvents—
Organic modifiers, such as methanol, acetonitrile, and others, are added to the aqueous buffer to increase the solubility of the solute or other additives and/or to affect the ionization degree of the sample components. The addition of these organic modifiers to the buffer generally causes a decrease in the electroosmotic flow.
Additives for Chiral Separations—
To separate optical isomers, a chiral selector is added to the separation buffer. The most commonly used chiral selectors are cyclodextrins, although in some cases crown ethers, certain polysaccharides, or even proteins can be used. Because chiral recognition is governed by the different interactions between the chiral selector and each of the enantiomers, the resolution achieved for the chiral compounds depends largely on the type of chiral selector used. While developing a given separation it may be useful to test cyclodextrins having a different cavity size (
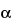
-,
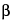
-, or
G-cyclodextrin) or modified cyclodextrins with neutral (methyl, ethyl, hydroxyalkyl, etc.) or ionizable (aminomethyl, carboxymethyl, sulfobutylether, etc.) moities. The resolution of chiral separations is also controlled by the concentration of the chiral selector, the composition and pH of the buffer, and the separation temperature. Organic additives, such as methanol or urea, can also affect the resolution of separation.
Capillary Gel Electrophoresis
Separation takes place inside a capillary filled with a polymer acting as a molecular sieve. The smaller components in the sample move faster along the capillary than the larger ones. This method can be used for separation of biopolymers-proteins, and DNA fragments, according to their molecular mass.
CHARACTERISTICS OF CHEMICAL and PHYSICAL GELS
Chemical Gels—
Chemical gels are prepared inside the capillary by reaction of monomers. One example of such a gel is a cross-linked polyacrylamide. This type of gel is bonded to the fused-silica wall and cannot be removed without destroying the capillary. For protein analysis, the separation buffer usually contains sodium dodecyl sulfate and the sample is denatured by heating in a mixture of sodium dodecyl sulfate and 2-mercaptoethanol or dithiothreitol before injection. Optimization of separation in a cross-linked gel is obtained by modifying the separation buffer (see Free Solution Capillary Electrophoresis) and by controlling the gel porosity during the gel preparation. For a cross-linked polyacrylamide gel, the porosity can be modified by changing the concentration of acrylamide and/or the ratio of the cross-linker. As a rule, a decrease in the porosity of the gel leads to a decrease in the mobility of the solutes. Due to the rigidity of this type of gel, only electrokinetic injection can be used.
Physical Gels—
Physical gels are hydrophilic polymers (i.e., linear polyacrylamide, cellulose derivatives, dextran, etc.) which can be dissolved in aqueous separation buffers, giving rise to a separation medium that also acts as a molecular sieve. These polymeric separation media are easier to prepare than cross-linked polymers. They can be prepared in a vial and filled by pressure in a wall-coated capillary with no electroosmotic flow. Replacing the gel before every injection generally improves the separation reproducibility. The porosity of the physical gels can be increased by using polymers of higher molecular weight (at a given polymer concentration) or by decreasing the polymer concentration (for a given polymer molecular weight). A decrease in gel porosity leads to a decrease in the mobility of the solute for the same buffer. Both hydrodynamic and electromigration injection techniques can be used, since the dissolution of these polymers in the buffer gives low viscosity solutions.
Capillary Isoelectric Focusing
The molecules migrate under the influence of the electric field, so long as they are charged, in a pH gradient generated by ampholytes having pI values in a wide range (poly-aminocarboxylic acids) dissolved in the separation buffer. The three basic steps in capillary isoelectric focusing are loading, focusing, and mobilization.
Loading—
Loading in One Step—
The sample is mixed with ampholytes and introduced into the capillary by pressure or vacuum.
Sequential Loading—
A leading buffer, then the ampholytes, then the sample mixed with ampholytes, again ampholytes alone, and finally the terminating buffer are introduced into the capillary. The volume of the sample must be small enough so as to not modify the pH gradient.
Focusing—
When the voltage is applied, ampholytes migrate toward the cathode or the anode according to their net charge, creating the pH gradient from anode (lower pH) to cathode (higher pH). The components to be separated migrate until they reach a pH corresponding to their isoelectric point and the current drops to very low values.
Mobilization—
The bands of separated components migrate past the detector by one of the three following methods.
Method 1—
During Focusing, under the influence of the electroosmotic flow when this flow is small enough to allow the focusing of the components.
Method 2—
By application of positive pressure after Focusing.
Method 3—
After
Focusing, by adding salts in the cathode reservoir or the anode reservoir (depending on the direction chosen for mobilization), in order to alter the pH in the capillary when the voltage is applied. As the pH is changed, the proteins and ampholytes are mobilized in the direction of the reservoir which contains added salts and pass the detector.
The separation achieved is expressed as DpI and depends on the pH gradient (dpH), the number of ampholytes having different pI values, the diffusion coefficient (D), the intensity of the electric field (E), and the variation of the electrophoretic mobility of the analyte with the pH, and is as follows:
in which dpH/dx is the pH gradient; and –dµ/dpH is the variation of the solution mobility with the pH in the region close to the pI.
Optimization Parameters—
The major parameters that need to be considered in the development of separations are voltage, capillary, and solutes.
Voltage—
Use of high fields from 300 V/cm to 1,000 V/cm during Focusing.
Capillary—
Depending on the Mobilization strategy selected (see above), the electroosmotic flow must be reduced or suppressed. Coated capillaries tend to reduce the electroosmotic flow.
Solutions—
The anode buffer reservoir is filled with a solution of a lower pH than the
pI of the most acidic ampholyte and the cathode reservoir is filled with a solution with a higher pH than the
pI of the most basic ampholyte. Phosphoric acid for the anode and sodium hydroxide for the cathode are frequently used.
Addition of a polymer, like methylcellulose, in the ampholyte solution tends to suppress convective forces (if any) and electroosmotic flow by increasing the viscosity. Commercial ampholytes covering many pH ranges are available and may also be mixed to obtain an expanded pH range. Broad pH ranges are used to estimate the isoelectric point whereas narrower ranges are employed to improve accuracy. Calibration can be made by correlating migration time with the isoelectric point of a series of standard protein markers. During Focusing, precipitation of proteins at their isoelectric point can be prevented, if necessary, using buffer additives such as glycerol, surfactants, urea, or Zwitterionic buffers. However, depending on the concentration, urea can denature proteins.
Micellar Electrokinetic Chromatography (MEKC)
Separation takes place in an electrolytic solution which contains a surfactant, generally ionic, at a concentration above the critical micellar concentration. The solute molecules are distributed between the aqueous buffer and the pseudo-stationary phase composed by the micelles according to the solute's partition coefficient. The technique can be considered as a hybrid of electrophoresis and chromatography. It is an electrophoretic technique that can be used for the separation of both neutral and charged solutes maintaining the efficiency, speed, and instrumental suitability of capillary electrophoresis. One of the most widely used surfactants is sodium dodecyl sulfate, although other anionic and cationic surfactants, such as cetyl trimethyl ammonium salts, have also been used.
At neutral and alkaline pH, a strong electroosmotic flow is generated and moves the separation buffer ions in the direction of the cathode. If sodium dodecyl sulfate is used as surfactant, the electrophoretic migration of the anionic micelle is in the opposite direction, toward the anode. As a result, the overall micelle migration velocity is slowed compared to the bulk flow of the electrolytic solution. In the case of neutral solutes, since the analyte can partition between the micelle and the aqueous buffer and has no electrophoretic mobility, the analyte migration velocity will only depend on the partition coefficient between the micelle and the aqueous buffer. In the electrophoretogram, the peak corresponding to each uncharged solute is always between that of the electroosmotic flow marker and that of the micelle, and the time elapsed between these two peaks is called the separation window. For electrically charged solutes, the migration velocity depends on both the partition coefficient of the solute between the micelle and the aqueous buffer and on the electrophoretic mobility of the solute in the absence of micelles.
The separation mechanism is essentially chromatographic, and migration of the solute and resolution can be expressed in terms of the capacity factor of the solute (K¢), which is the ratio between the total number of moles of solute in the micelle to those in the mobile phase. For a neutral compound, K¢ is as follows:
in which tr is the migration time of the solute; to is the analysis time of the unretained solute obtained by injecting an electroosmotic flow marker which does not enter the micelle (i.e., methanol); tm is the micelle migration time measured by injecting a micelle marker, such as Sudan III, which migrates continuously associated in the micelle; K is the partition coefficient of the solute; VS is the volume of the micelles phase; and VM is the volume of the mobile phase.
The resolution between two closely-migrating compounds (RS) is as follows:
in which
N is the number of theoretical plates for one of the compounds;
is the selectivity obtained;
K¢a and
K¢b are capacity factors for both components; and the other terms are as defined above.
Similar, but not identical, equations give K¢ and RS values for electrically charged compounds.
Optimization Parameters—
The main parameters to be considered in the development of separations by MEKC are instrumental and electrolytic solution parameters.
INSTRUMENTAL PARAMETERS
Voltage—
Separation time is inversely proportional to applied voltage. An increase in voltage can cause excessive heat production that gives rise to temperature gradients and viscosity gradients of the buffer in the cross section of the capillary. This effect can be significant with high conductivity buffers, such as those containing micelles. Poor heat dissipation causes band broadening and decreases resolution.
Temperature—
Variations in capillary temperature affect the partition coefficient of the solute between the buffer and the micelle, the critical micelle concentration, and the viscosity of the buffer. These parameters contribute to the migration time of the solutes.
Capillary—
Length and internal diameter contribute to analysis time and efficiency of separations. Increasing both effective length and total length can decrease the electrical fields, working at constant voltage, and will increase migration time and improve the separation efficiency. The internal diameter controls heat dissipation, at a given buffer and electrical field, and provides a broadening of the sample band.
ELECTROLYTIC SOLUTION PARAMETERS
Surfactant Type and Concentration—
The type of surfactant, as the stationary phase in chromatography, affects the resolution since it modifies separation selectively. The log
K¢ of a neutral compound increases linearly with the concentration of detergent in the mobile phase. Resolution in MEKC reaches a maximum when
K¢ approaches the value of
modifying the concentration of surfactant in the mobile phase changes the resolution.
Buffer pH—
pH does not modify the partition coefficient of non-ionized solutes, but it can modify the electroosmotic flow in uncoated capillaries. A decrease in the buffer pH decreases the electroosmotic flow and therefore increases the resolution of the neutral solutes, giving rise to longer analysis time.
Organic Solvents—
To improve separation of hydrophobic compounds, organic modifiers (methanol, propanol, acetonitrile, etc.) can be added to the separation electrolytic solution. The addition of these modifiers generally decreases migration time and selectivity of the separation. The addition of organic modifiers affects micelle formation, thus a given surfactant concentration can be used only with a certain percentage of organic modifier before the micellezation equilibrium is eliminated or adversely affected, resulting in the absence of micelles and therefore the absence of the partition mechanism of MEKC. The elimination of micelles in the presence of a high content of organic solvent does not always mean that the separation will no longer be possible, since in some cases, the hydrophobic interaction between the ionic surfactant monomer and the neutral solutes form solvophobic complexes that can be separated electrophoretically.
Additives for Chiral Separations—
A chiral selector is included in the micellar system, either covalently bound to the surfactant or added to the micellar separation electrolyte. Micelles which have a moiety with chiral discrimination properties include salts, N-dodecanoyl-L-amino acids, bile salts, etc. Chiral resolution can also be achieved using chiral discriminators, such as cyclodextrins added to the electrolytic solutions which contain micelliced achiral surfactants.
Other Additives—
Selectivity can be modified by adding chemicals to the buffer. Addition of several types of cyclodextrins to the buffer are also used to reduce the interaction of hydrophobic solutes with the micelle, increasing the selectivity for this type of compound. The addition of substances able to modify solute-micelle interactions by adsorption on the latter, has been used to improve the selectivity of the separations in MEKC. These additives may consist of a second surfactant (ionic or non-ionic) which gives rise to mixed micelles, metallic cations which dissolve in the micelle, and give co-ordination complexes with the solutes.
QUANTITATIVE ANALYSIS
Peak areas must be divided by the corresponding migration time to give the corrected area in order to compensate for the shift in migration time from run to run, thus reducing the variation of the response. It will also compensate for the different responses of sample constituents with different migration times. Where an internal standard is used, check that no peak of the substance to be examined is masked by that of the internal standard.
Calculations—
From the values obtained, calculate the content of a component or components being determined. When indicated, the percentage of one (or more) components of the sample to be examined is calculated by determining the areas of the peak(s) as a percentage of the total corrected areas of all the peaks, excluding those due to solvents or any added reagents. The use of an automatic integration system (integrator or data acquisition and processing system) is recommended.
Capillary Electrophoresis System Suitability
The choice of suitability parameters to be used will depend on the type of capillary electrophoresis that is performed. These parameters are the capacity factor (K¢) used only for Micelles Electrokinetic Chromatography, the number of theoretical plates (n), the symmetry factor (AS), and the resolution (RS). Note that in previous sections, the theoretical expression for n and RS have been described, but more practical equations that allow for the determination of these suitability parameters using the electrophoretograms are described below.
The number of theoretical plates (n) may be calculated from the formula:
n = 5.54 (t / b0.5)2,
in which t is the distance, in mm, along the baseline between the point of injection and the perpendicular dropped from the maximum of the peak in question; and b0.5 is the peak width, in mm, at half height.
The resolution (RS) may be calculated from the formula:
RS = 1.18(
tb 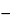 ta / b0.5b
ta / b0.5b +
b0.5a),
in which tb and ta are the distances, in mm, along the baseline, between the point of injection and the perpendicular dropped from the maxima of two adjacent peaks (tb > ta); and b0.5b and b0.5a are the peak widths, in mm, at half height.
The resolution (
RS) may be also calculated by measuring the height of the valley (
c) between two partly resolved peaks in a standard preparation, the height of the smaller peak (
d), and by specifying (
c/d)
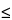 x
x, in which
x is the limit indicated in the individual monograph.
The symmetry factor of a peak (AS) may be calculated using the formula:
AS = b0.05/2A,
in which b0.05 is the width of the peak at one-twentieth of the peak height; and A is the distance between the perpendicular dropped from the peak maximum and the leading edge of the peak at one-twentieth of the peak height.
Other system suitability parameters include tests for area repeatability (i.e., standard deviation of areas or of area/migration time) and tests for migration time repeatability (i.e., standard deviation of migration time). For migration time repeatability, it will be necessary to provide for a test to measure the suitability of the capillary washing procedures. To avoid the lack of repeatability of the migration time, an alternative practice is to use a migration time relative to an internal standard.
A test for the verification of the signal-to-noise ratio for a standard preparation or the determination of the limit of quantitation is a useful system suitability parameter. The detection limit and quantitation limit correspond to a signal-to-noise ratio greater than 3 and 10, respectively. The signal-to-noise ratio (S/N) is calculated as follows:
S/N = 2H/hn,
in which H is the height of the peak corresponding to the component concerned in the electrophoretogram obtained with the specified reference solution; and hn is the absolute value of the largest noise fluctuation from the baseline in an electrophoretogram obtained after injection of a blank and observed over a distance equal to twenty times the width at the half-height of the peak in the electrophoretogram obtained with the reference solution, and situated equally around the place where this peak would be found.
;)
;)
;)
;)
;)
;)
;)
;)
;)