Drug release  724
724 —
—
Test 1:
If the product complies with this test, the labeling indicates that it meets USP
Drug Release Test 1.
Medium:
Phosphoric acid solution (1 in 1000); 250 mL, in a tall-form beaker.
Apparatus 7—
Proceed as directed in the chapter, using the transdermal system holder–cylinder (see
Figure 7b). Center the Transdermal System onto a dry, unused 10-cm × 10-cm piece of Cuprophan dialysis membrane with the adhesive side against the membrane, taking care to eliminate air bubbles between the membrane and the release surface. Attach the membrane to the cylinder using two Parker O-rings, such that one of the borders of the transdermal system is aligned to the groove and it is wrapped around the cylinder. The filled beakers are weighed and pre-equilibrated to 32.0 ± 0.3
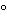
, prior to immersing the test sample. Reciprocate at a frequency of about 30 cycles per minute with an amplitude of 2.0 ± 0.1 cm. At the end of each time interval, transfer the test sample to a fresh beaker containing the appropriate volume of
Medium, weighed and pre-equilibrated to 32.0 ± 0.3
. At the end of each release interval, allow the beakers to cool to room temperature, make up for evaporative losses by adding water to obtain the original weight, and mix. This solution is the final
Test solution.
Times:
2, 12, and 24 hours.
Determine the amount of C10H14N2 released by employing the following method.
Mobile phase—
Transfer 0.2 mL of N,N-dimethyloctylamine to a 1-L volumetric flask, add 220 mL of acetonitrile, and mix. Add 300 mL of water, 0.2 mL of glacial acetic acid, 0.20 g of anhydrous sodium acetate, and 0.55 g of sodium 1-dodecanesulfonate, and dilute with water to volume. Mix for 1 hour until clear. Filter and degas. Make adjustments if necessary (see
System Suitability under
Chromatography 621). (Equilibration of the column may take as long as 3 hours.)
Standard solution—
Dissolve an accurately weighed quantity of
USP Nicotine Bitartrate Dihydrate RS in
Medium, and dilute quantitatively, and stepwise if necessary, with
Medium to obtain a solution having a known concentration of about 0.142 mg of nicotine bitartrate per mL (or 0.046 mg nicotine as free base per mL).
[NOTE—About 80 mL of this solution is required in order to prepare the
System suitability solution.]
System suitability solution—
Transfer 8 mg (free base) of nicotine to a 100-mL volumetric flask, and dissolve in 10 mL of acetonitrile. Add 5 mL of 30 percent hydrogen peroxide, and allow 15 minutes to react. Dilute with Medium to volume, and mix. Transfer 20 mL of this solution to a 100-mL volumetric flask, dilute with Standard solution to volume, and mix.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 254-nm detector and a 4.6-mm × 15-cm column that contains packing L1. The flow rate is about 1 mL per minute. Chromatograph the
System suitability solution, and record the peak responses as directed for
Procedure: the resolution,
R, between nicotine and any degradation peaks is not less than 1.1; the tailing factor is not more than 2.0; and the relative standard deviation for replicate injections is not more than 1.5%.
Procedure—
Separately inject equal volumes (about 50 µL) of filtered portions of the Standard solution and the solution under test into the chromatograph, record the chromatograms, and measure the responses for the major peaks.
Tolerances—
The amount of C
10H
14N
2 released, as a percentage of the labeled amount of the dose absorbed in vivo, at the times specified below, conforms to
Acceptance Table 4.
| Time (hours) |
Amount dissolved |
| 0–2 |
between 31% and 87% |
| 2–12 |
between 62% and 191% |
| 12–24 |
between 85% and 261% |
Test 2:
If the product complies with this test, the labeling indicates that it meets USP
Drug Release Test 2.
Phosphate buffer—
Dissolve 40.0 g of sodium chloride, 1.0 g of potassium chloride, 8.66 g of dibasic sodium phosphate, and 1.0 g of monobasic potassium phosphate in 5 L of water.
Medium:
Phosphate buffer; 500 mL.
Apparatus 6:
50 rpm, double-sided tape being used to attach the Transdermal System to the cylinder.
Times:
6 and 24 hours.
Determine the amount of C10H14N2 released by employing the following method.
Mobile phase—
Proceed as directed in the Assay.
System suitability solution—
Transfer 1.0 mL of the System suitability solution, prepared as directed in the Assay, to a 100-mL volumetric flask, dilute with Medium to volume, and mix.
Standard solution—
Pipet 6.0 mL of the Standard preparation, prepared as directed in the Assay, into a 50-mL volumetric flask, dilute with Medium to volume, and mix. Dilute quantitatively and stepwise with Medium to obtain an appropriate final concentration.
Test solution—
At each of the test times, withdraw a 2-mL aliquot of the solution under test. [NOTE—Replace the aliquots withdrawn for analysis with fresh portions of Medium.]
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 260-nm detector and a 4.6-mm × 12.5-cm column that contains packing L1. The flow rate is about 1 mL per minute. Chromatograph the
Standard solution used for the 6-hour interval, and record the peak responses as directed for
Procedure: the resolution,
R, between 4,4
¢-dipyridyl dihydrochloride and nicotine is not less than 5.0; the tailing factor is not more than 2.0; and the relative standard deviation for replicate injections is not more than 2.0%.
Procedure—
Separately inject equal volumes (about 100 µL) of the filtered portion of the Standard solution and the Test solution into the chromatograph, record the chromatograms, and measure the responses for the major peaks.
Tolerances—
The amount of C
10H
14N
2 released, as a percentage of the labeled amount of the dose absorbed in vivo, at the times specified, conforms to
Acceptance Table 4.
| Time (hours) |
Amount dissolved |
| 6 |
between 71% and 157% |
| 24 |
between 156% and 224% |
Test 3:
If the product complies with this test, the labeling indicates that it meets USP
Drug Release Test 3.
Medium:
water; 900 mL.
Apparatus 5:
50 rpm, the stainless steel disk assembly being replaced with a 5-cm watch glass for an 11-mg Transdermal System and an 8-cm watch glass for a 22-mg Transdermal System.
Times:
1, 2, and 4 hours.
Standard solution—
Prepare a solution of USP Nicotine Bitartrate RS in water having a known concentration of nicotine similar to that of the solution under test.
Procedure—
Determine the amount of C10H14N2 released by employing UV absorption at the wavelength of maximum absorbance at about 259 nm, in comparison with the Standard solution, using water as the blank.
Tolerances—
The amount of C
10H
14N
2 released, as a percentage of the labeled amount of the dose absorbed in vivo, at the times specified, conforms to the following
Acceptance Table.
| Time (hours) |
Amount dissolved |
| 1 |
between 35% and 75% |
| 2 |
between 55% and 95% |
| 4 |
not less than 73% |
Acceptance Table
| Level |
Tested |
Criteria |
| L1 |
6 |
No individual value lies outside each of the stated ranges and no individual value is less than the stated amount at the final test time. |
| L2 |
6 |
The average value of the 12 units (L1 + L2) lies within each of the stated ranges and is not less than the stated amount at the final test time; none is more than 5% of the labeled content outside each of the stated ranges; and none is more than 5% of the labeled content below the stated amount at the final test time. |
| L3 |
12 |
The average value of the 24 units (L1 + L2 + L3) lies within each of the stated ranges and is not less than the stated amount at the final test time; not more than 2 of the 24 units are more than 5% of labeled content outside each of the stated ranges; not more than 2 of the 24 units are more than 5% of the labeled content below the stated amount at the final test time; and none of the units is more than 10% of the labeled content outside each of the stated ranges or more than 10% of the labeled content below the stated amount at the final test time. |
Test 4:
If the product complies with this test, the labeling indicates that it meets USP
Drug Release Test 4.
Medium:
0.025 N hydrochloric acid; 600 mL.
Apparatus 5:
50 rpm, a convex screen being used to hold the Transdermal System in position during testing.
Times:
4 and 16 hours.
Standard solution and Procedure—
Proceed as directed for Procedure in Test 3.
Tolerances—
The amount of C
10H
14N
2 released, as a percentage of the labeled amount of the dose absorbed in vivo, at the times specified, conforms to
Acceptance Table 4.
| Time (hours) |
 Amount dissolved Amount dissolved |
| 4 |
between 36% and 66% |
| 16 |
between 72% and 112% |
Test 5:
If the product complies with this test, the labeling indicates that it meets USP
Drug Release Test 5.
Phosphate buffer, Medium, and Apparatus—
Proceed as directed under Test 2.
Times:
3, 6, and 24 hours.
Mobile phase—
Proceed as directed in the Assay.
System suitability solution, Standard solution, Test solution, and Chromatographic system—
Proceed as directed under Test 2.
Procedure—
Proceed as directed under Test 2 except to inject about 30 µL.
Tolerances—
The amount of C
10H
14N
2 released, as a percentage of the labeled amount of the dose absorbed in vivo, at the times specified, conforms to
Acceptance Table 5.
| Time (hours) |
Amount dissolved |
| 3 |
between 79% and 112% |
| 6 |
between 108% and 141% |
| 24 |
between 156% and 202% |
Drug release 724—
Test 1—
If the product complies with this test, the labeling indicates that it meets USP
Drug Release Test 1.
Medium:
Phosphoric acid solution (1 in 1000); 250 mL, in a tall-form beaker.
Apparatus 7—
Proceed as directed in the chapter, using the transdermal system holder–cylinder (see
Figure 4b). Center the Transdermal System onto a dry, unused 10-cm × 10-cm piece of Cuprophan dialysis membrane with the adhesive side against the membrane, taking care to eliminate air bubbles between the membrane and the release surface. Attach the membrane to the cylinder using two Parker O-rings, such that one of the borders of the transdermal system is aligned to the groove and it is wrapped around the cylinder. The filled beakers are weighed and pre-equilibrated to 32.0 ± 0.3
, prior to immersing the test sample. Reciprocate at a frequency of about 30 cycles per minute with an amplitude of 2.0 ± 0.1 cm. At the end of each time interval, transfer the test sample to a fresh beaker containing the appropriate volume of
Medium, weighed and pre-equilibrated to 32.0 ± 0.3
. At the end of each release interval, allow the beakers to cool to room temperature, make up for evaporative losses by adding water to obtain the original weight, and mix. This solution is the final
Test solution.
Times:
2, 12, and 24 hours.
Determine the amount of C10H14N2 released by employing the following method.
Mobile phase—
Transfer 0.2 mL of
N,N-dimethyloctylamine to a 1-L volumetric flask, add 220 mL of acetonitrile, and mix. Add 300 mL of water, 0.2 mL of glacial acetic acid, 0.20 g of anhydrous sodium acetate, and 0.55 g of sodium 1-dodecanesulfonate, and dilute with water to volume. Mix for 1 hour until clear. Filter and degas. Make adjustments if necessary (see
System Suitability under
Chromatography 621).
[Note—Equilibration of the column may take as long as 3 hours.
]
Standard solution—
Dissolve an accurately weighed quantity of
USP Nicotine Bitartrate Dihydrate RS in
Medium, and dilute quantitatively, and stepwise if necessary, with
Medium to obtain a solution having a known concentration of about 0.142 mg of nicotine bitartrate per mL (or 0.046 mg nicotine as free base per mL).
[NOTE—About 80 mL of this solution is required in order to prepare the
System suitability solution.]
System suitability solution—
Transfer 8 mg (free base) of nicotine to a 100-mL volumetric flask, and dissolve in 10 mL of acetonitrile. Add 5 mL of 30 percent hydrogen peroxide, and allow 15 minutes to react. Dilute with Medium to volume, and mix. Transfer 20 mL of this solution to a 100-mL volumetric flask, dilute with Standard solution to volume, and mix.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 254-nm detector and a 4.6-mm × 15-cm column that contains packing L1. The flow rate is about 1 mL per minute. Chromatograph the
System suitability solution, and record the peak responses as directed for
Procedure: the resolution,
R, between nicotine and any degradation peaks is not less than 1.1; the tailing factor is not more than 2.0; and the relative standard deviation for replicate injections is not more than 1.5%.
Procedure—
Separately inject equal volumes (about 50 µL) of filtered portions of the Standard solution and the solution under test into the chromatograph, record the chromatograms, and measure the responses for the major peaks.
Tolerances—
The amount of C
10H
14N
2 released, as a percentage of the labeled amount of the dose absorbed in vivo, at the times specified below, conforms to
Acceptance Table 1.
| Time (hours) |
Amount dissolved |
| 0–2 |
between 31% and 87% |
| 2–12 |
between 62% and 191% |
| 12–24 |
between 85% and 261% |
Test 2—
If the product complies with this test, the labeling indicates that it meets USP
Drug Release Test 2.
Phosphate buffer—
Dissolve 40.0 g of sodium chloride, 1.0 g of potassium chloride, 8.66 g of dibasic sodium phosphate, and 1.0 g of monobasic potassium phosphate in 5 L of water.
Medium:
Phosphate buffer; 500 mL.
Apparatus 6:
50 rpm, double-sided tape being used to attach the Transdermal System to the cylinder.
Times:
6 and 24 hours.
Determine the amount of C10H14N2 released by employing the following method.
Mobile phase—
Proceed as directed in the Assay.
System suitability solution—
Transfer 1.0 mL of the System suitability solution, prepared as directed in the Assay, to a 100-mL volumetric flask, dilute with Medium to volume, and mix.
Standard solution—
Pipet 6.0 mL of the Standard preparation, prepared as directed in the Assay, into a 50-mL volumetric flask, dilute with Medium to volume, and mix. Dilute quantitatively and stepwise with Medium to obtain an appropriate final concentration.
Test solution—
At each of the test times, withdraw a 2-mL aliquot of the solution under test. [NOTE—Replace the aliquots withdrawn for analysis with fresh portions of Medium.]
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 260-nm detector and a 4.6-mm × 12.5-cm column that contains packing L1. The flow rate is about 1 mL per minute. Chromatograph the
Standard solution used for the 6-hour interval, and record the peak responses as directed for
Procedure: the resolution,
R, between 4,4
¢-dipyridyl dihydrochloride and nicotine is not less than 5.0; the tailing factor is not more than 2.0; and the relative standard deviation for replicate injections is not more than 2.0%.
Procedure—
Separately inject equal volumes (about 100 µL) of the filtered portion of the Standard solution and the Test solution into the chromatograph, record the chromatograms, and measure the responses for the major peaks.
Tolerances—
The amount of C
10H
14N
2 released, as a percentage of the labeled amount of the dose absorbed in vivo, at the times specified, conforms to
Acceptance Table 1.
| Time (hours) |
Amount dissolved |
| 6 |
between 71% and 157% |
| 24 |
between 156% and 224% |
Test 3—
If the product complies with this test, the labeling indicates that it meets USP
Drug Release Test 3.
Medium:
water; 900 mL.
Apparatus 5:
50 rpm, the stainless steel disk assembly being replaced with a 5-cm watch glass for an 11-mg Transdermal System and an 8-cm watch glass for a 22-mg Transdermal System.
Times:
1, 2, and 4 hours.
Standard solution—
Prepare a solution of USP Nicotine Bitartrate RS in water having a known concentration of nicotine similar to that of the solution under test.
Procedure—
Determine the amount of C10H14N2 released by employing UV absorption at the wavelength of maximum absorbance at about 259 nm, in comparison with the Standard solution, using water as the blank.
Tolerances—
The amount of C
10H
14N
2 released, as a percentage of the labeled amount of the dose absorbed in vivo, at the times specified, conforms to the following
Acceptance Table.
| Time (hours) |
Amount dissolved |
| 1 |
between 35% and 75% |
| 2 |
between 55% and 95% |
| 4 |
not less than 73% |
Acceptance Table
| Level |
Tested |
Criteria |
| L1 |
6 |
No individual value lies outside each of the stated ranges and no individual value is less than the stated amount at the final test time. |
| L2 |
6 |
The average value of the 12 units (L1 + L2) lies within each of the stated ranges and is not less than the stated amount at the final test time; none is more than 5% of the labeled content outside each of the stated ranges; and none is more than 5% of the labeled content below the stated amount at the final test time. |
| L3 |
12 |
The average value of the 24 units (L1 + L2 + L3) lies within each of the stated ranges and is not less than the stated amount at the final test time; not more than 2 of the 24 units are more than 5% of labeled content outside each of the stated ranges; not more than 2 of the 24 units are more than 5% of the labeled content below the stated amount at the final test time; and none of the units is more than 10% of the labeled content outside each of the stated ranges or more than 10% of the labeled content below the stated amount at the final test time. |
Test 4—
If the product complies with this test, the labeling indicates that it meets USP
Drug Release Test 4.
Medium:
0.025 N hydrochloric acid; 600 mL.
Apparatus 5:
50 rpm, a convex screen being used to hold the Transdermal System in position during testing.
Times:
4 and 16 hours.
Standard solution and Procedure—
Proceed as directed under Test 3.
Tolerances—
The amount of C
10H
14N
2 released, as a percentage of the labeled amount of the dose absorbed in vivo, at the times specified, conforms to
Acceptance Table 1.
| Time (hours) |
Amount dissolved |
| 4 |
between 36% and 66% |
| 16 |
between 72% and 112% |
Test 5—
If the product complies with this test, the labeling indicates that it meets USP
Drug Release Test 5.
Phosphate buffer, Medium, and Apparatus—
Proceed as directed under Test 2.
Times:
3, 6, and 24 hours.
Mobile phase—
Proceed as directed in the Assay.
System suitability solution, Standard solution, Test solution, and Chromatographic system—
Proceed as directed under Test 2.
Procedure—
Proceed as directed under Test 2 except to inject about 30 µL.
Tolerances—
The amount of C
10H
14N
2 released, as a percentage of the labeled amount of the dose absorbed in vivo, at the times specified, conforms to
Acceptance Table 1.
| Time (hours) |
Amount dissolved |
| 3 |
between 79% and 112% |
| 6 |
between 108% and 141% |
| 24 |
between 156% and 202% |
(Official April 1, 2006)
Assay—
Mobile phase—
Mix 300 mL of acetonitrile, 700 mL of water, and 1 mL of triethylamine, filter, and degas. Make adjustments if necessary (see
System Suitability under
Chromatography 621).
Standard preparation—
Dissolve an accurately weighed quantity of
USP Nicotine Bitartrate Dihydrate RS in water to obtain a stock solution having a known concentration of about 26.87 mg per mL. Quantitatively dilute a volume of the stock solution with methanol to obtain a solution having a known concentration of about 5.37 mg of
USP Nicotine Bitartrate Dihydrate RS per mL.
[NOTE—This solution contains 1.75 mg of nicotine per mL.
]
System suitability solution—
Transfer an accurately weighed quantity of about 12.5 mg of 4,4¢-dipyridyl dihydrochloride to a 25-mL volumetric flask, add 5.0 mL of the Standard preparation, dilute with methanol to volume, and mix.
Assay preparation—
Cut an accurately counted number of Transdermal Systems, equivalent to about 175 mg of nicotine, into strips 5 cm2 in area. Remove the protective liners, if any, from the strips, and discard. Transfer the strips to a 250-mL flask, and add 100.0 mL of methanol. Insert the stopper into the flask, and shake by mechanical means for about 3 hours. Filter, and use the clear filtrate as the Assay preparation.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 260-nm detector and a 4.6-mm × 25-cm column containing packing L1. The flow rate is about 1.5 mL per minute. Chromatograph the
System suitability solution, and record the peak responses as directed for
Procedure: the resolution,
R, between nicotine and 4,4
¢-dipyridyl dihydrochloride is not less than 5.0. Chromatograph the
Standard preparation, and record the peak responses as directed for
Procedure: the relative standard deviation for replicate injections is not more than 1.0%.
Procedure—
Separately inject equal volumes (about 10 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the areas for the major peaks. Calculate the quantity, in mg, of nicotine (C
10H
14N
2) in each Transdermal System taken by the formula:
(162.23/462.41)(100C/N)(rU / rS),
in which 162.23 and 462.41 are the molecular weights of nicotine and anhydrous nicotine bitartrate, respectively;
C is the concentration, in mg per mL, of
USP Nicotine Bitartrate Dihydrate RS in the
Standard preparation; N is the number of Nicotine Transdermal Systems taken for the
Assay preparation; and
rU and
rS are the nicotine peak responses obtained from the
Assay preparation and the
Standard preparation, respectively.