All high-risk level CSPs for administration by injection into the vascular and central nervous systems that are prepared in groups of more than 25 identical individual single-dose packages (such as ampuls, bags, syringes, and vials), or in multiple dose vials for administration to multiple patients, or are exposed longer than 12 hours at 2
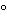
to 8
and longer than 6 hours at warmer than 8
before they are sterilized are tested to ensure that they are sterile (see
Sterility Tests  71
71
) and do not contain excessive bacterial endotoxins (see
Bacterial Endotoxins Test 85). All CSPs that are intended to be solutions must be visually examined for the presence of particulate matter and not administered or dispensed when such matter is observed. The prescription orders, written compounding procedure, preparation records, and expended materials used to make CSPs in all contamination risk levels are inspected for accuracy of correct identities and amounts of ingredients, aseptic mixing and sterilization, packaging, labeling, and expected physical appearance before they are administered or dispensed.