The process of thermal sterilization employing saturated steam under pressure, or autoclaving, is the preferred method to terminally sterilize aqueous preparations that have been verified to maintain their full chemical and physical stability under the conditions employed (see
Steam Sterilization under
Sterilization and Sterility Assurance of Compendial Articles  1211
1211
). To achieve sterility, it is necessary that all materials be exposed to steam at 121
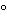
, under a pressure of about one atmosphere or 15 psi, for the duration verified by testing to achieve sterility of the items, which is usually 20 to 60 minutes for CSPs. An allowance must be made for the time required for the material to reach 121
before the sterilization exposure duration is timed.