Recommended Protocol for Conducting a Single-Dose Bioequivalence Study under Fasting Conditions—
Objective—
The objective is to compare the rate and extent of absorption of a generic formulation with that of the reference formulation when given as equal labeled doses.
Design—
The study design is a single-dose, two-treatment, two-period, two-sequence, crossover study with a washout period of at least 4 months. An equal number of subjects should be randomly assigned to the two possible dosing sequences. As an alternative, the design may be a single-dose, randomized, two-treatment, one-period, parallel study with a blood sampling time of approximately 3 months. Before the study begins, the proposed protocol should be approved by an institutional review board.
Facilities—
The clinical facilities and analytical laboratory used for the study should be identified along with the names, titles, and curriculum vitae of medical and scientific or analytical directors.
Selection of Subjects—
The sponsor should enroll a number of subjects sufficient to ensure statistical validity of the study. It is recommended that a minimum of 48 subjects be used for crossover studies or 96 subjects for parallel studies. Subjects should be healthy, preferably nonsmoking, volunteers aged 18 to 50 years and within 10% of ideal body weight for height and build, although within 15% of ideal body weight is acceptable (Metropolitan Life Insurance Company Statistical Bulletin, 1983). Subjects should be selected on the basis of acceptable medical history, physical examination, and clinical laboratory test results. Female subjects must be given a pregnancy test prior to beginning the study. Subjects with any current or past medical condition that might significantly affect their pharmacokinetic or pharmacodynamic response to the administered drug should be excluded from the study. If smokers are included, they should be identified as such. Written, informed consent must be obtained from all study participants before they are accepted into the study.
Procedure—
Following an overnight fast of at least 10 hours, subjects should be administered a single dose of the test or reference product with 240 mL of water.
Restrictions—
Study volunteers should observe the following restrictions:
-
The subjects should fast for 5 hours post-dose. Water may be taken except for 2 hours after drug administration.
-
No xanthine-containing foods or beverages should be consumed for 48 hours prior to dosing and until 24 hours post-dose.
-
No alcoholic beverages should be consumed for 48 hours prior to and during the study period.
-
Subjects should take no Rx medications, including oral contraceptives, beginning 2 weeks and no OTC medications beginning 1 week before drug administration and until after the study is completed.
Blood Sampling—
Venous blood samples should be collected into tubes containing an anti-coagulant pre-dose (0 hours), and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48, 72, 96, 120 hours and at 8, 12, 15, 19, 22, 26, 29, 33, 36, 43, 50, 57, 64, 70, 80, and 90 days. Blood should be immediately frozen until assayed.
Subject Monitoring—
Blood pressure and pulse rate should be monitored hourly during the first 4 hours of the study. Subjects with a heart rate less than 45 bpm or greater than 110 bpm should have an electrocardiogram (lead II) performed and have their pulse monitored hourly. Subjects should report any unusual symptoms observed during the study. Subjects should be periodically questioned during each phase of the study for any unusual symptoms experienced after drug administration. A physical examination and clinical laboratory tests should be repeated after completing the blood level study.
Analytical Methods—
Hydroxychloroquine in whole blood should be assayed using a suitable method fully validated with respect to adequate sensitivity, specificity, linearity, recovery, accuracy, and precision (both within and between days). The method to be used should be specific enough to measure the parent drug without interferences from metabolites and endogenous or exogenous components in the blood. Stability of samples under frozen conditions, at room temperature, and during freeze-thaw cycles, if appropriate, should be determined. Chromatograms of the analysis of the unknown samples, including all associated standard curves and quality control chromatograms, should be available for regulatory authorities. The sponsor should justify the rejection of any analytical data and provide a rationale for selection of the reported values.
Pharmacokinetic Analysis—
From the blood drug concentration-time data, the following pharmacokinetic parameters should be determined for hydroxychloroquine:
-
Area under the concentration-time curve from time zero to time t (AUC0–t), calculated by the trapezoidal rule, where t is the last time point with measurable non-zero concentration.
-
Area under the concentration-time curve from time zero to time infinity (AUC
0–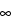
), where AUC
0– = AUC
0–t +
Ct /(
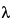 z
z),
Ct is the last measurable non-zero concentration, and
z is the terminal elimination rate constant calculated using an appropriate method.
-
The terminal phase elimination rate constant,
z, and the elimination half-life are calculated using an appropriate method.
-
Peak drug concentration, Cmax, and the time to peak drug concentration, Tmax, are obtained directly from the data without interpolation.
Statistical Analysis of Pharmacokinetic Data—
See Statistical Procedures for Bioequivalence Studies Using a Standard Two-Treatment, Crossover Design under General Guidances; and an appropriate model should be chosen for the parallel study.
;)
;)
;)
;)