TOTAL PROTEIN ASSAY
The following procedures are provided as illustrations of the determination of total protein content in pharmacopeial preparations. Other techniques, such as HPLC, are also acceptable if total protein recovery is demonstrated. Many of the total protein assay methods described below can be performed successfully using kits from commercial sources. [NOTE—Where water is required, use distilled water.]
Method 1
Protein in solution absorbs UV light at a wavelength of 280 nm, due to the presence of aromatic amino acids, mainly tyrosine and tryptophan. This property is the basis of this method. Protein determination at 280 nm is mainly a function of the tyrosine and tryptophan content of the protein. If the buffer used to dissolve the protein has a high absorbance relative to that of water, there is an interfering substance in the buffer. This interference can be compensated for when the spectrophotometer is adjusted to zero buffer absorbance. If the interference results in a large absorbance that challenges the limit of sensitivity of the spectrophotometer, the results may be compromised. Furthermore, at low concentrations protein can be absorbed onto the cuvette, thereby reducing the content in solution. This can be prevented by preparing samples at higher concentrations or by using a nonionic detergent in the preparation. [NOTE—Keep the Test Solution, the Standard Solution, and the buffer at the same temperature during testing.]
Test Solution—
Dissolve a suitable quantity of the protein under test in the appropriate buffer to obtain a solution having a concentration of 0.2 to 2 mg per mL.
Standard Solution—
Unless otherwise specified in the individual monograph prepare a solution of USP Reference Standard or reference material for the protein under test in the same buffer and at the same concentration as the Test Solution.
Procedure—
Concomitantly determine the absorbances of the
Standard Solution and the
Test Solution in quartz cells at a wavelength of 280 nm, with a suitable spectrophotometer (see
Spectrophotometry and Light-Scattering  851
851
), using the buffer as the blank. To obtain accurate results, the response should be linear in the range of protein concentrations to be assayed.
Light-Scattering—
The accuracy of the UV spectroscopic determination of protein can be decreased by the scattering of light by the test specimen. If the proteins in solution exist as particles comparable in size to the wavelength of the measuring light (250 to 300 nm), scattering of the light beam results in an apparent increase in absorbance of the test specimen. To calculate the absorbance at 280 nm due to light-scattering, determine the absorbances of the Test Solution at wavelengths of 320, 325, 330, 335, 340, 345, and 350 nm. Using the linear regression method, plot the log of the observed absorbance versus the log of the wavelength, and determine the standard curve best fitting the plotted points. From the graph so obtained, extrapolate the absorbance value due to light-scattering at 280 nm. Subtract the absorbance from light-scattering from the total absorbance at 280 nm to obtain the absorbance value of the protein in solution. Filtration with a filter having a 0.2-µm porosity or clarification by centrifugation may be performed to reduce the effect of light-scattering, especially if the solution is noticeably turbid.
Calculations—
Calculate the concentration,
CU, of protein in the test specimen by the formula:
CS(AU / AS),
in which
CS is the concentration of the
Standard Solution; and
AU and
AS are the corrected absorbances of the
Test Solution and the
Standard Solution, respectively (see
Spectrophotometry and Light-Scattering 851).
Method 2
This method, commonly referred to as the Lowry assay, is based on the reduction by protein of the phosphomolybdic-tungstic mixed acid chromogen in the Folin-Ciocalteu's phenol reagent, resulting in an absorbance maximum at 750 nm. The Folin-Ciocalteu's phenol reagent reacts primarily with tyrosine residues in the protein, which can lead to variation in the response of the assay to different proteins. Because the method is sensitive to interfering substances, a procedure for precipitation of the protein from the test specimen may be used. Where separation of interfering substances from the protein in the test specimen is necessary, proceed as directed below for Interfering Substances prior to preparation of the Test Solution. The effect of interfering substances can be minimized by dilution, provided the concentration of the protein under test remains sufficient for accurate measurement.
Standard Solutions—
Unless otherwise specified in the individual monograph, dissolve the USP Reference Standard or reference material for the protein under test in the buffer used to prepare the Test Solution. Dilute portions of this solution with the same buffer to obtain not fewer than five Standard Solutions having concentrations between 5 and 100 µg of protein per mL, the concentrations being evenly spaced.
Test Solution—
Dissolve a suitable quantity of the protein under test in the appropriate buffer to obtain a solution having a concentration within the range of the concentrations of the Standard Solutions. An appropriate buffer will produce a pH in the range of 10.0 to 10.5.
Blank—
Use the buffer used for the Test Solution and the Standard Solutions.
Reagents and Solutions—
Copper Sulfate Reagent—
Dissolve 100 mg of cupric sulfate and 200 mg of sodium tartrate in water, dilute with water to 50 mL, and mix. Dissolve 10 g of sodium carbonate in water to a final volume of 50 mL, and mix. Slowly pour the sodium carbonate solution into the copper sulfate solution with mixing. Prepare this solution fresh daily.
SDS Solution—
Dissolve 5 g of sodium dodecyl sulfate in water, and dilute with water to 100 mL.
Sodium Hydroxide Solution—
Dissolve 3.2 g of sodium hydroxide in water, dilute with water to 100 mL, and mix.
Alkaline Copper Reagent—
Prepare a mixture of Copper Sulfate Reagent, SDS Solution, and Sodium Hydroxide Solution (1:2:1). This reagent may be stored at room temperature for up to 2 weeks.
Diluted Folin-Ciocalteu’s Phenol Reagent—
Mix 10 mL of Folin-Ciocalteu’s phenol TS with 50 mL of water. Store in an amber bottle, at room temperature.
Procedure—
To 1 mL of each
Standard Solution, the
Test Solution, and the
Blank, add 1 mL of
Alkaline Copper Reagent, and mix. Allow to stand at room temperature for 10 minutes. Add 0.5 mL of the
Diluted Folin-Ciocalteu's Phenol Reagent to each solution, mix each tube immediately, and allow to stand at room temperature for 30 minutes. Determine the absorbances of the solutions from the
Standard Solutions and the
Test Solution at the wavelength of maximum absorbance at 750 nm, with a suitable spectrophotometer (see
Spectrophotometry and Light-Scattering 851), using the solution from the
Blank to set the instrument to zero.
Calculations—
[NOTE—The relationship of absorbance to protein concentration is nonlinear; however, if the standard curve concentration range is sufficiently small, it will approach linearity.] Using the linear regression method, plot the absorbances of the solutions from the Standard Solutions versus the protein concentrations, and determine the standard curve best fitting the plotted points. From the standard curve so obtained and the absorbance of the Test Solution, determine the concentration of protein in the Test Solution.
INTERFERING SUBSTANCES
In the following procedure, deoxycholate-trichloroacetic acid is added to a test specimen to remove interfering substances by precipitation of proteins before testing. This technique also can be used to concentrate proteins from a dilute solution.
Sodium Deoxycholate Reagent—
Prepare a solution of sodium deoxycholate in water having a concentration of 150 mg in 100 mL.
Trichloroacetic Acid Reagent—
Prepare a solution of trichloroacetic acid in water having a concentration of 72 g in 100 mL.
Procedure—
Add 0.1 mL of
Sodium Deoxycholate Reagent to 1 mL of a solution of the protein under test. Mix on a vortex mixer, and allow to stand at room temperature for 10 minutes. Add 0.1 mL of
Trichloroacetic Acid Reagent, and mix on a vortex mixer. Centrifuge at 3000 ×
g for 30 minutes, decant the liquid, and remove any residual liquid with a pipet. Redissolve the protein pellet in 1 mL of
Alkaline Copper Reagent. Proceed as directed for the
Test Solution.
NOTE—Color development reaches a maximum in 20 to 30 minutes during incubation at room temperature, after which there is a gradual loss of color. Most interfering substances cause a lower color yield; however, some detergents cause a slight increase in color. A high salt concentration may cause a precipitate to form. Because different protein species may give different color response intensities, the standard protein and test protein should be the same.
Method 3
This method, commonly referred to as the Bradford assay, is based on the absorption shift from 470 nm to 595 nm observed when the brilliant blue G dye binds to protein. The brilliant blue G dye binds most readily to arginyl and lysyl residues in the protein, which can lead to variation in the response of the assay to different proteins.
Standard Solutions—
Unless otherwise specified in the individual monograph, dissolve the USP Reference Standard or reference material for the protein under test in the buffer used to prepare the Test Solution. Dilute portions of this solution with the same buffer to obtain not fewer than five Standard Solutions having concentrations between 100 µg and 1 mg of protein per mL, the concentrations being evenly spaced.
Test Solution—
Dissolve a suitable quantity of the protein under test in the appropriate buffer to obtain a solution having a concentration within the range of the concentrations of the Standard Solutions.
Blank—
Use the buffer used to prepare the Test Solution and the Standard Solutions.
Coomassie Reagent—
Dissolve 100 mg of brilliant blue G
3 in 50 mL of alcohol.
[NOTE—Not all dyes have the same brilliant blue G content, and different products may give different results.
] Add 100 mL of phosphoric acid, dilute with water to 1 L, and mix. Pass the solution through filter paper (Whatman No. 1 or equivalent), and store the filtered reagent in an amber bottle at room temperature.
[NOTE—Slow precipitation of the dye will occur during storage of the reagent. Filter the reagent before use.
]
Procedure—
Add 5 mL of the
Coomassie Reagent to 100 µL of each
Standard Solution, the
Test Solution, and the
Blank, and mix by inversion. Avoid foaming, which will lead to poor reproducibility. Determine the absorbances of the solutions from the
Standard Solutions and the
Test Solution at 595 nm, with a suitable spectrophotometer (see
Spectrophotometry and Light-Scattering 851), using the
Blank to set the instrument to zero.
[NOTE—Do not use quartz (silica) spectrophotometer cells: the dye binds to this material. Because different protein species may give different color response intensities, the standard protein and test protein should be the same.
]
There are relatively few interfering substances, but detergents and ampholytes in the test specimen should be avoided. Highly alkaline specimens may interfere with the acidic reagent.
Calculations—
[NOTE—The relationship of absorbance to protein concentration is nonlinear; however, if the standard curve concentration range is sufficiently small, it will approach linearity.] Using the linear regression method, plot the absorbances of the solutions from the Standard Solutions versus the protein concentrations, and determine the standard curve best fitting the plotted points. From the standard curve so obtained and the absorbance of the Test Solution, determine the concentration of protein in the Test Solution.
Method 4
This method, commonly referred to as the bicinchoninic acid or BCA assay, is based on reduction of the cupric (Cu2+) ion to cuprous (Cu1+) ion by protein. The bicinchoninic acid reagent is used to detect the cuprous ion. The method has few interfering substances. When interfering substances are present, their effect may be minimized by dilution, provided that the concentration of the protein under test remains sufficient for accurate measurement.
Standard Solutions—
Unless otherwise specified in the individual monograph, dissolve the USP Reference Standard or reference material for the protein under test in the buffer used to prepare the Test Solution. Dilute portions of this solution with the same buffer to obtain not fewer than five Standard Solutions having concentrations between 10 and 1200 µg of protein per mL, the concentrations being evenly spaced.
Test Solution—
Dissolve a suitable quantity of the protein under test in the appropriate buffer to obtain a solution having a concentration within the range of the concentrations of the Standard Solutions.
Blank—
Use the buffer used to prepare the Test Solution and the Standard Solutions.
Reagents—
BCA Reagent—
Dissolve about 10 g of bicinchoninic acid, 20 g of sodium carbonate monohydrate, 1.6 g of sodium tartrate, 4 g of sodium hydroxide, and 9.5 g of sodium bicarbonate in water. Adjust, if necessary, with sodium hydroxide or sodium bicarbonate to a pH of 11.25. Dilute with water to 1 L, and mix.
Copper Sulfate Reagent—
Dissolve about 2 g of cupric sulfate in water to a final volume of 50 mL.
Copper-BCA Reagent—
Mix 1 mL of Copper Sulfate Reagent and 50 mL of BCA Reagent.
Procedure—
Mix 0.1 mL of each
Standard Solution, the
Test Solution, and the
Blank with 2 mL of the
Copper-BCA Reagent. Incubate the solutions at 37
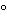
for 30 minutes, note the time, and allow to come to room temperature. Within 60 minutes following the incubation time, determine the absorbances of the solutions from the
Standard Solutions and the
Test Solution in quartz cells at 562 nm, with a suitable spectrophotometer (see
Spectrophotometry and Light-Scattering 851), using the
Blank to set the instrument to zero. After the solutions are cooled to room temperature, the color intensity continues to increase gradually. If substances that will cause interference in the test are present, proceed as directed for
Interfering Substances under
Method 2. Because different protein species may give different color response intensities, the standard protein and test protein should be the same.
Calculations—
[NOTE—The relationship of absorbance to protein concentration is nonlinear; however, if the standard curve concentration range is sufficiently small, it will approach linearity.] Using the linear regression method, plot the absorbances of the solutions from the Standard Solutions versus the protein concentrations, and determine the standard curve best fitting the plotted points. From the standard curve so obtained and the absorbance of the Test Solution, determine the concentration of protein in the Test Solution.
Method 5
This method, commonly referred to as the Biuret assay, is based on the interaction of cupric (Cu2+) ion with protein in an alkaline solution and the resultant development of absorbance at 545 nm.
Standard Solutions—
Unless otherwise specified in the individual monograph, prepare a solution of Albumin Human for which the protein content has been previously determined by nitrogen analysis (using the nitrogen-to-protein conversion factor of 6.25) or of the USP Reference Standard or reference material for the protein under test in sodium chloride solution (9 in 1000). Dilute portions of this solution with sodium chloride solution (9 in 1000) to obtain not fewer than three Standard Solutions having concentrations between 0.5 and 10 mg per mL, the concentrations being evenly spaced. [NOTE—Low responses may be observed if the sample under test has significantly different level of proline than that of Albumin Human. A different standard protein may be employed in such cases.]
Test Solution—
Prepare a solution of the test protein in sodium chloride solution (9 in 1000) having a concentration within the range of the concentrations of the Standard Solutions.
Blank—
Use sodium chloride solution (9 in 1000).
Biuret Reagent—
Dissolve about 3.46 g of cupric sulfate in 10 mL of hot water, and allow to cool (Solution 1). Dissolve about 34.6 g of sodium citrate dihydrate and 20.0 g of sodium carbonate in 80 mL of hot water, and allow to cool (Solution 2). Mix Solution 1 and Solution 2, and dilute with water to 200 mL. This Biuret Reagent is stable at room temperature for 6 months. Do not use the reagent if it develops turbidity or contains any precipitate.
Procedure—
To one volume of a solution of the
Test Solution add an equal volume of sodium hydroxide solution (6 in 100), and mix. Immediately add a volume of
Biuret Reagent equivalent to 0.4 volume of the
Test Solution, and mix. Allow to stand at a temperature between 15
and 25
for not less than 15 minutes. Within 90 minutes after the addition of the
Biuret Reagent, determine the absorbances of the
Standard Solutions and the solution from the
Test Solution at the wavelength of maximum absorbance at 545 nm, with a suitable spectrophotometer (see
Spectrophotometry and Light-Scattering 851), using the
Blank to set the instrument to zero.
[NOTE—Any solution that develops turbidity or a precipitate is not acceptable for calculation of protein concentration.
]
Calculations—
Using the least-squares linear regression method, plot the absorbances of the Standard Solutions versus the protein concentrations, determine the standard curve best fitting the plotted points, and calculate the correlation coefficient for the line. [NOTE—Within the given range of the standards, the relationship of absorbance to protein concentration is approximately linear.] A suitable system is one that yields a line having a correlation coefficient of not less than 0.99. From the standard curve and the absorbance of the Test Solution, determine the concentration of protein in the test specimen, making any necessary correction.
Interfering Substances—
To minimize the effect of interfering substances, the protein can be precipitated from the initial test specimen as follows. Add 0.1 volume of 50 percent trichloroacetic acid to 1 volume of a solution of the test specimen, withdraw the supernatant layer, and dissolve the precipitate in a small volume of 0.5 N sodium hydroxide. Use the solution so obtained to prepare the Test Solution.
Comments—
This test shows minimal difference between equivalent IgG and albumin samples. Addition of the sodium hydroxide and the Biuret Reagent as a combined reagent, insufficient mixing after the addition of the sodium hydroxide, or an extended time between the addition of the sodium hydroxide solution and the addition of the Biuret Reagent will give IgG samples a higher response than albumin samples. The trichloroacetic acid method used to minimize the effects of interfering substances can also be used to determine the protein content in test specimens at concentrations below 500 µg per mL.
Method 6
This fluorometric method is based on the derivatization of the protein with
o-phthalaldehyde (OPA), which reacts with the primary amines of the protein (i.e., NH
2-terminal amino acid and the
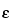
-amino group of the lysine residues). The sensitivity of the test can be increased by hydrolyzing the protein before testing. Hydrolysis makes the
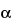
-amino group of the constituent amino acids of the protein available for reaction with the
o-phthalaldehyde reagent. The method requires very small quantities of the protein.
Primary amines, such as tris(hydroxymethyl)aminomethane and amino acid buffers, react with o-phthalaldehyde and must be avoided or removed. Ammonia at high concentrations will react with o-phthalaldehyde as well. The fluorescence obtained when amine reacts with o-phthalaldehyde can be unstable. The use of automated procedures to standardize this procedure may improve the accuracy and precision of the test.
Standard Solutions—
Unless otherwise specified in the individual monograph, dissolve the USP Reference Standard or reference material for the protein under test in the buffer used to prepare the Test Solution. Dilute portions of this solution with the same buffer to obtain not fewer than five Standard Solutions having concentrations between 10 and 200 µg of protein per mL, the concentrations being evenly spaced.
Test Solution—
Dissolve a suitable quantity of the protein under test in the appropriate buffer to obtain a solution having a concentration within the range of the concentrations of the Standard Solutions.
Blank—
Use the buffer used to prepare the Test Solution and the Standard Solutions.
Reagents—
Borate Buffer—
Dissolve about 61.83 g of boric acid in water, and adjust with potassium hydroxide to a pH of 10.4. Dilute with water to 1 L, and mix.
Stock OPA Reagent—
Dissolve about 120 mg of o-phthalaldehyde in 1.5 mL of methanol, add 100 mL of Borate Buffer, and mix. Add 0.6 mL of polyoxyethylene (23) lauryl ether, and mix. This solution is stable at room temperature for at least 3 weeks.
OPA Reagent—
To 5 mL of Stock OPA Reagent add 15 µL of 2-mercaptoethanol. Prepare at least 30 minutes prior to use. This reagent is stable for one day.
Procedure—
Adjust each of the
Standard Solutions and the
Test Solution to a pH between 8 and 10.5. Mix 10 µL of the
Test Solution and each of the
Standard Solutions with 100 µL of
OPA Reagent, and allow to stand at room temperature for 15 minutes. Add 3 mL of 0.5 N sodium hydroxide, and mix. Using a suitable fluorometer (see
Spectrophotometry and Light-Scattering 851), determine the fluorescent intensities of solutions from the
Standard Solutions and the
Test Solution at an excitation wavelength of 340 nm and an emission wavelength between 440 and 455 nm.
[NOTE—The fluorescence of an individual specimen is read only once because irradiation decreases the fluorescent intensity.
]
Calculations—
The relationship of fluorescence to protein concentration is linear. Using the linear regression method, plot the fluorescent intensities of the solutions from the Standard Solutions versus the protein concentrations, and determine the standard curve best fitting the plotted points. From the standard curve so obtained and the fluorescent intensity of the Test Solution, determine the concentration of protein in the test specimen.
Method 7
This method is based on nitrogen analysis as a means of protein determination. Interference caused by the presence of other nitrogen-containing substances in the test specimen can affect the determination of protein by this method. Nitrogen analysis techniques destroy the protein under test but are not limited to protein presentation in an aqueous environment.
Procedure 1—
Determine the nitrogen content of the protein under test as directed under
Nitrogen Determination 461. Commercial instrumentation is available for the Kjeldahl nitrogen assay.
Procedure 2—
Commercial instrumentation is available for nitrogen analysis. Most nitrogen analysis instruments use pyrolysis (i.e., combustion of the sample in oxygen at temperatures approaching 1000
), which produces nitric oxide (NO) and similar oxides of nitrogen (NO
x) from the nitrogen present in the test protein. Some instruments convert the nitric oxides to nitrogen gas, which is quantified with a thermal conductivity detector. Other instruments mix nitric oxide (NO) with ozone (O
3) to produce excited nitrogen dioxide (NO
2), which emits light when it decays and can be quantified with a chemiluminescence detector. A protein reference material or reference standard that is relatively pure and is similar in composition to the test proteins is used to optimize the injection and pyrolysis parameters and to evaluate consistency in the analysis.
Calculations—
The protein concentration is calculated by dividing the nitrogen content of the sample by the known nitrogen content of the protein. The known nitrogen content of the protein can be determined from the chemical composition of the protein or by comparison with the nitrogen content of the USP Reference Standard or reference material.
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)