PROTEIN SEQUENCING
Protein sequencing is useful in the control of quality of protein biologicals because it can provide primary structure information, i.e., amino terminal and/or carboxy terminal structure. For rDNA-derived biologicals, this methodology has the additional purpose of confirming the complementary DNA (cDNA)-predicted amino acid sequence, protein homogeneity, and the potential extent of proteolytic clips. For monoclonal antibodies, this technique is used for determining protein homogeneity. Protein sequencing is divided into amino-terminal and carboxy-terminal sequencing applications and procedures.
Amino-Terminal Sequencing—
Amino-terminal sequence analysis is a classical protein chemistry technique that yields significant information about primary structure (sequence), homogeneity, and the presence of known or unknown cleavages in the polypeptide. N-terminal sequence analysis is performed with a number of commercially available automatic peptide sequencers. The method is based on the coupling reaction of the amino terminal residue of a protein or peptide with PITC. The resulting PTC-amino acid derivative is cleaved from the protein by a perfluoridated organic acid (generally trifluoroacetic or heptafluorobutyric acid), which exposes the adjacent amino acid. This next amino acid serves as a new N-terminus and is derivatized in the subsequent coupling and cleavage cycle. This process is repeated until an appropriate number, normally 8 to 10, of the amino acids are removed. The modified amino acid residue resulting from the cleavage cycle (anilinothiazolinone [ATZ]) is generally converted in the presence of acid and heat to a phenylthiohydantoin-amino acid (PTH-AA). The PTH-AA may then be determined following RP-HPLC analysis. Any intrachain cleavages as well as heterogeneity of the N-termini (e.g., N-terminal methionine) on the polypeptide will also be sequenced at the same time. These result in smaller peaks in the chromatogram and may enable both the relative quantitation of the amount of the N-termini and the identification of the location of the cleavage site on the polypeptide. This procedure for protein sequence analysis may also be performed manually. The limitations of the PITC sequencing method are that the method is only semiquantitative (i.e., the amount of the N-termini can only be estimated) and the PTH derivatives of serine and threonine may be severely degraded, making their determination difficult. Cysteine residues in order to be determined, must first be modified, for example by alkylation. In addition, the amino acids glycine and proline are slow to rearrange, resulting in minor difficulty in their determination.
Carboxy-Terminal Sequencing—
Sequencing of the protein from the carboxy terminus also yields valuable primary structure information as well as possible C-terminal cleavages. The sequential degradation of a protein from the C-terminus can be performed by either chemical or enzymatic methods. The reaction of hydrazine, ammonium thiocyanate, or cyanogen bromide with a protein can be used to sequentially degrade the protein at or near the C-terminus. The ammonium thiocyanate reaction has been extended for use on proteins coupled to solid supports. The C-terminal amino acids can be sequentially cleaved enzymatically with exopeptidases such as carboxypeptidases. Limitations of the carboxypeptidase approach are the potential contamination with endopeptidase and the inherent difficulty and unpredictable nature of the sequencing. Mass spectrometry can be used either directly on protein digests or in conjunction with HPLC peptide mapping to identify the C-terminus of the protein. However, these methods are only semiquantitative.
PEPTIDE MAPPING BY HPLC
For pharmaceutical proteins, peptide mapping has two primary purposes: it is a highly specific identity method and, in the case of biotechnology-derived products, may serve as a confirmation of genetic stability. Peptide mapping is used to compare the protein structure of a specific lot of material to that of a suitable reference material/reference standard or to those structures of previous lots to confirm correctness of the primary structure and to confirm lot-to-lot consistency of primary structure (within the limits of this technique). The amino- and carboxy-terminal peptides and carbohydrate-containing peptides often can be separated and identified. The latter are valuable in the peptide maps of glycosylated proteins such as monoclonal antibodies. Peptide mapping may be used to determine the presence of single or multiple incorrect amino acids that may result from such events as a single point mutation or mistranslation of the cDNA sequence.
The procedure involves the selective fragmentation of the protein into discrete peptides that are resolved by some chromatographic technique. The fragmentation is accomplished with endoproteases, such as trypsin, chymotrypsin, thermolysin, or V8 protease, or by selective chemical degradation with cyanogen bromide, which cleaves at specific sites on the molecule. Selection of the appropriate endoprotease to be used is directed by the primary sequence of the protein. Trypsin cleaves on the C-terminal side of the basic residues lysine and arginine; chymotrypsin cleaves after the aromatic residues phenylalanine, tyrosine, and tryptophan; thermolysin cleaves after the hydrophobic residues leucine, isoleucine, and valine; V8 protease cleaves after the acidic residues glutamic acid and aspartic acid; and cyanogen bromide cleaves at methionyl residues. Other enzymes, such as clostripain (arginine) and endoproteinase lys-C (lysine), and chemical methods, such as 2-nitro-5-thiocyanobenzoic acid (cysteine), may also be used. Each of these methods has its own set of advantages and disadvantages. One common disadvantage to all these techniques is that nonspecific cleavages occur to some degree. It is important that the peptides generated from the digestion are large enough to provide structural information about the protein, and yet small enough to allow their analysis and separation by a technique such as RP-HPLC. For this reason and the fact that cleavage with this enzyme is almost quantitative, trypsin is the enzyme with the most general applicability for most proteins. For large proteins of greater than 60,000 daltons (about 520 amino acids), cleavage with trypsin may result in too many fragments, so another endoprotease may be chosen. L-TPCK (tosyl-L-phenylalanine chloromethyl ketone)-treated trypsin normally is used because TPCK inhibits the action of chymotrypsin, a contaminant present in many trypsin preparations. Although reaction with cyanogen bromide cleaves at methionyl residues, proteins do not contain many of these residues. As a result, relatively few peptides are obtained and these may be too large or hydrophobic for HPLC separation.
Once the digestion is complete, the peptides are generally separated by either RP-HPLC and/or HPIEC. Selection of the appropriate column is empirically based and will vary for different proteins. For RP-HPLC, both 100-
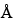
and 300-
pore size supports work well, and the selection of the silica support may be an important criterion for optimal separation. For the smaller peptides generated by these digestions, C8 and C18 stationary phases generally have been found to be more efficient than C4 supports. The most common solvents used for reversed-phase separations are water and acetonitrile containing a constant (
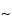
0.1%) amount of trifluoroacetic acid. Buffered mobile phases containing phosphate also offer excellent selectivity depending on the pH. Screening the effect of pH in the 3.0 to 5.0 range causes a shift of peptides containing the acidic residues, glutamic acid and aspartic acid. For ion-exchange separations, less information is available, but both silica and polymeric supports with both weak and strong ion-exchange stationary support can be used successfully. Because many of the peptides are somewhat hydrophobic, the addition of small amounts of organic solvents in the mobile phase, such as 5% to 10% methanol or acetonitrile, may be necessary. A potential disadvantage of HPIEC analysis of peptide mixtures is that sometimes neutral peptides or peptides that have the same charge as the support may not be retained on the column and thus may not be separated or identified by this method.
IMMUNOASSAYS
Immunoassays are used either as active drug substance methods to identify and quantitate the protein of interest or as impurity profile methods to detect and quantitate known host cell protein impurities. Because these protein impurities may represent a large number of potential impurities at trace levels rather than a single impurity, the immunoassays must be sensitive and selective to detect as many of these impurities as possible. Immunoassays that can measure these impurities to very low levels have been developed for E. coli proteins (ECPs) and CHO proteins. Immunoassays additionally may serve as potency assays for monoclonal antibodies using an appropriate antigen.
Immunoassays consist of a large group of assays that depend on specific high-affinity antibody:antigen interactions. These assays include the radioimmunoassays (RIAs) and the enzyme-linked immunosorbent assays (ELISAs). RIAs are performed in a liquid or solid phase using an unlabeled antibody directed against the radiolabeled protein of interest. The principle of the RIA is that the inhibition of binding of labeled antigen to unlabeled antibody by samples is compared to the inhibition by known standards, thus allowing quantitation of the protein of interest.
Immunoradiometric assays (IRMAs) or sandwich RIAs employ two antibody preparations that are used to sandwich the protein of interest. The first antibody is unlabeled and is directed against the protein, and the second antibody is radiolabeled and may be directed against the protein or the first antibody. The entire antibody:antigen complex is isolated and the amount of radioactivity, and, therefore, the protein of interest, is determined. The development of an RIA or IRMA for a biotechnology-derived product requires careful attention to production of the antisera, preparation of the labeled tracer, preparation of a suitable reference standard, and methods for the separation of free antigen from bound antigen.
The most commonly used ELISA format of trace impurity analysis is the sandwich ELISA that utilizes two antibody preparations like the IRMA, but without radiolabeling. The first antibody is unlabeled and the second antibody has an enzyme such as horseradish peroxidase (HRP) or alkaline phosphatase attached. Basically, the ELISA method consists of applying a layer of purified antibodies to the host cell proteins onto microtitration plates, followed by the protein product. The enzyme antibody conjugate is added and allowed to bind to the antibody-bound host cell proteins. An appropriate substrate is added for color development, which is analyzed with a spectrophotometric plate reader. Such a multiantigen ELISA requires a representative reference standard preparation of appropriate host cell protein impurities to serve as the immunogen for preparation of the antibodies used for the assay. This reference standard preparation is usually prepared from a manufacturing production run yielding all of the expected host cell proteins except the product protein. The total absence of the product protein is necessary in this preparation to avoid the production of antibodies to the product itself when the reference standard is used as an immunogen. Because of varying affinities of polyclonal antibodies to multiantigen preparations, the absolute accuracy of the multiantigen methods and the ability to detect every potential antigen cannot be guaranteed.
ELECTROPHORESIS
Electrophoretic assays are among the most common and powerful of the assays used to evaluate protein purity and homogeneity. They are valuable not only for the initial evaluation and release of biotechnology-derived products but also as stability-indicating methods for detecting molecular or chemical changes in the molecule as a result of denaturation, aggregation, oxidation, deamidation, etc. The use of these methods is facilitated by their simplicity and their requirement of only microgram quantities of sample. The two types of electrophoretic assays most often used for biotechnology-derived products are sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and isoelectric focusing (IEF).
The SDS-PAGE method separates proteins primarily by their molecular weight because, in the presence of the anionic detergent SDS, a net negatively charged protein SDS complex is formed. The sample is first denatured in the detergent, which disrupts the noncovalent intramolecular and intermolecular bonds that hold proteins together, and then it is electrophoresed through a polyacrylamide gel support. Protein migration through the gel is proportional to size so that smaller proteins migrate faster through the gel than larger ones. Samples are often electrophoresed under both reduced and nonreduced conditions to determine if impurities of the same molecular weight or if intramolecular proteolytic cleavages of the protein of interest are present. Although nonreducing SDS-PAGE is commonly used to estimate the state of aggregation and/or oligomerization of the protein of interest, this method will only permit observation of aggregates or oligomers that are stable in the presence of SDS and the conditions used for sample preparation and electrophoresis. Proteins consisting of multiple chains held together by disulfide bonds are broken down and separated into their individual polypeptide chains. Sample detection following electrophoresis can be quantitative with densitometric analysis of Coomassie Brilliant Blue stain or qualitative, but with increased sensitivity in the nanogram range, with silver stain. Silver stain SDS-PAGE may also be performed quantitatively under suitable conditions. With proper validation, Coomassie Brilliant Blue staining and densitometry may also be used to give quantitative determination of polypeptides in the nanogram range. SDS-PAGE coupled with Coomassie Brilliant Blue stain is used to quantitatively determine the purity of the sample with regard to dimer and larger covalent aggregates and fragments. When the method is combined with the silver stain technique, an assessment of low/trace levels of a new impurity can be made by directly comparing the electrophoresed sample to the electrophoresed reference material or standard under reduced and nonreduced conditions. Generally, silver staining is used qualitatively because of potential major variations in binding of silver from protein to protein, and relatively inconsistent background on a routine basis. An estimation of the quantity of an impurity can be obtained by electrophoresis of a known amount of an internal standard such as bovine serum albumin or lesser dilutions of the protein of interest on other lanes of the same gel. The SDS-PAGE separation of a protein can be combined with an immunological method such as immunoblotting. The resulting Western blot is used to determine the identity of an electrophoretic band (i.e., product related or host cell protein impurity). After electrophoresis, the separated proteins are transferred onto a nitrocellulose or polyvinylidene difluoride (PVDF) membrane and reacted with the antibody of interest. Visualization of the complex is done with an enzymatically or radiolabeled antibody.
The IEF method separates proteins on the basis of their charge in an electrical field. The charges on a protein originate from various sources within its amino acid composition, such as protonated amino groups, unprotonated carboxyl groups, deprotonated sulfhydryl groups or tyrosine residues, oxidized cysteine residues, and deamidated residues. However, for each protein there is a pH at which the protein is isoelectric and these charges cancel each other, with the net charge being effectively zero. IEF is performed in the native state in a support of loose-pore polyacrylamide or agarose gel containing ampholytes (amphoteric low molecular weight ions) that set up a pH gradient because of their migration within the support matrix when an electrical field is applied. Simultaneously, in the presence of the electrical field, positively charged proteins migrate towards the cathode and negatively charged proteins migrate towards the anode. Migration stops when each protein reaches the pH value in the support gradient where its net charge is zero. This is the apparent pI or isoelectric point of the protein. Because the migration of a protein is dependent on its amino acid composition, altered forms of the protein and other proteins will migrate to different points on the support. IEF gels may be stained for protein visualization with either Coomassie Brilliant Blue or silver stains. IEF is employed as an identity tool or to ensure the homogeneity of a protein (e.g., monoclonal antibodies) as demonstrated by a banding pattern with the correct pI range. The method can also be used to evaluate the stability of a biological product. Protein deamidation (i.e., glutamine or asparagine residue deamidation) over time leading to the production of a new carboxylic acid group results in molecules with a more acidic pI. IEF can provide information on the state of glycosylation of glycoproteins such as monoclonal antibodies, which may appear as many bands because of changes in the apparent charge on the protein molecule as a result of the sialic acid residues. IEF gel patterns are usually more complicated to interpret than those of SDS-PAGE and interpretation may require many assumptions or subjective judgment.
High-performance capillary electrophoresis (HPCE), which offers the potential advantages of very high protein resolution, is being thoroughly investigated because of recent advances in the technology.
CHROMATOGRAPHIC METHODS
Chromatographic methods have long been used in the determination of the purity of small organic molecules and proteins such as insulin (see
Chromatography  621
621
) and in the determination of the active ingredient and/or excipient concentration of pharmaceutical products. Chromatographic methods are also very effective in the determination of the purity of recombinant pharmaceuticals. However, the chromatography of proteins is far more difficult because of multiple modes of interaction with the chromatographic support as a result of the size and/or shape, charge, and hydrophobicity of the proteins. The most common chromatographic methods used to profile recombinant proteins are RP-HPLC, HPIEC, size-exclusion chromatography (HPSEC), and hydrophobic interaction chromatography (HIC). These methods involve the separation of proteins and are used to determine the purity of drug substances as well as the levels of known impurities or degradation products. A complication with all column chromatographic methods is determining the mass balance between column load and column eluate. Nevertheless, HPLC techniques are valuable for determining the purity and strength of protein pharmaceuticals.
The most common RP-HPLC analyses are performed on columns containing a C4 or C8 stationary phase on a silica-based or polymeric support. C18 stationary phases are also used but more often with smaller peptides in an application such as peptide mapping. Supports with pore sizes of at least 300
are preferred for proteins of molecular weight greater than about 10,000. For most RP-HPLC analyses, the proteins are eluted with aqueous acetonitrile gradients and the trifluoroacetic acid is kept constant at 0.1%. Other buffers such as phosphate or tris(hydroxymethyl)aminomethane (Tris) are also used, where the pH may be adjusted for added selectivity to achieve optimal separations.
HPIEC is an important method for purity determination. These separations are based on changes in the charge of the molecule and are useful for identifying and quantitating in protein pharmaceuticals common impurities such as oxidized (primarily oxidized methionine) and deamidated forms (glutamine and asparagine) and clipped or truncated forms. Both strong and weak ion-exchange stationary phases on either silica or polymeric supports can be used. Cation-exchange chromatography may be performed on sulfopropyl-type resins and are effective in distinguishing oxidation and deamidation products. Proteins are typically loaded onto a column equilibrated with water or a weak buffer, and eluted with a salt gradient, such as 0 to 1 M sodium chloride.
HPSEC is a technique that may provide information on the levels of aggregation and fragmentation in a protein pharmaceutical. Depending on the information needed, the mobile phase may be native, containing an aqueous buffer such as 100 mM phosphate, pH 7, or it may be denaturing, containing a low level of a chaotrope or detergent such as 0.1% SDS. The analyses are performed isocratically, with detection typically between 210 and 220 nm depending on the buffer used. Detection at 280 nm may also be used but is less sensitive. Classical size-exclusion chromatography was performed on soft polymeric supports such as cross-linked dextrans, polyacrylamide, or agarose. These, however, are better suited for low-pressure applications. As a result, a number of supports with increased mechanical strength have been developed. Commercially available silica-based and cross-linked agarose supports are now commonly used. HPSEC is also useful for the determination of clipped forms of proteins. Clipped chains often remain attached through the disulfide bonds of cysteine residues. Treatment of the sample with a reducing agent such as dithiothreitol or mercaptoethanol will cleave the disulfide bond and separate the chains. The clipped chains may then be resolved from unclipped forms by HPSEC.
HIC provides separation of proteins based on differences in their hydrophobicity under mild adsorption and elution conditions that generally prevent denaturation and subsequent loss of biological activity. A stationary phase that is weakly hydrophobic is used with a buffered aqueous mobile phase and an initial high-salt concentration to adsorb the protein, which is then selectively eluted using a decreasing salt gradient. Interactions occur between nonpolar amino acid residues that are exposed on the surface of the protein and hydrophobic groups that are present on the chromatographic matrix. A number of silica-based and polymeric supports combined with weakly hydrophobic ligands, such as polyethers, phenyl ethers, or short alkyl chains, have been developed for use in HIC. This technique can be used in the analysis, purification, and characterization of more labile hydrophobic proteins. Protein retention and selectivity can be modified by control of variables such as salt type and concentration, pH and selective-ion effects, temperature and gradient design, as well as by careful selection of the stationary phase.
QUANTITATIVE ASSAYS
Biomimetic assays (assays that mimic the biological effect of the product) are of major significance in the discussion of assays for biotechnology-derived products. These assays measure the activity of the product and ensure that it is efficacious. Essentially, there are three major types of quantitative assays: animal model assays, cell culture-based assays and in-vitro (physicochemical) assays. Each of these assays has application in the control of biological products. Regardless of the type of quantitative assay employed, it is desirable and, in some cases, necessary, to use a biomimetic assay.
Animal Model Assays—
Biomimetic assays in animal models have been developed for routine use. Although these assays have a relatively long history of use, they have several major disadvantages such as the large number of animals and appropriate animal facilities and handlers required, the high cost of analysis, the long analysis time (i.e., several days to weeks), and poor reproducibility of results. They are, however, in use mainly because a cell culture-based or in-vitro assay has not been developed and demonstrated to be of equal or greater value. An example of such an assay is that used for the determination of the activity of human growth hormone (somatrem and somatropin). The potency of human growth hormone is determined with a rat weight gain bioassay. Hypophysectomized female rats are monitored for weight gain over an 11-day period after daily injections with human growth hormone. The relative potency of the test sample is obtained by statistical comparison of the activity of the sample to that of a reference material/reference standard. Animal models can be used as bioidentity tests if and when appropriate in-vitro biological and/or physicochemical assays are developed for the measurement of potency of products.
Cell Culture-Based Bioassays—
This group of assays is comparatively easier to perform, gives results faster (1 to 3 days), and is considerably less expensive and less wasteful of resources than the animal model assays. Cell culture-based bioassays provide information on the effect of the biological product in a living system, but they are imprecise as a consequence of the variances of living cells but not as imprecise as an animal model assay. However, they can be automated and therefore can be repeated sufficiently to provide relatively reproducible and accurate results. An example of this type of assay is the measurement of antiviral activity of human
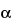
-interferon in a human diploid foreskin cell line or in a human lung carcinoma cell line (A549). This assay is performed in microtitration plates by incubation of cells with
-interferon and subsequent challenge with encephalomyocarditis virus. The cells that survive are detected by dye binding and the dilution of
-interferon where 50% protection of the monolayer occurs is calculated.
In Vitro (Physicochemical) Assays—
This group of assays does not rely on a living model, but is usually based on the chemical action of a biological product. These methods are comparatively simple, fast, precise, and accurate. The activity of tissue-type plasminogen activator (alteplase), for example, can be determined with an in vitro clot lysis assay that can be automated and can provide the required results within hours. A synthetic fibrin clot is formed in the presence of plasminogen as a result of the action of the enzyme thrombin on fibrinogen. When alteplase is added, the plasminogen is converted to the active enzyme plasmin, which then lyses the synthetic clot. The assay endpoint is followed spectrophotometrically or visually by noting the release of entrapped air bubbles. Another advantage of this type of assay, because of its precision and accuracy, is that it can be used to provide reliable estimates of the stability of the product. Examples of antibody:antigen and protein:ligand (receptor)-based in-vitro bioassays have also been developed for specific applications. These types of assays offer many advantages in their application to determine the potency of monoclonal antibodies or other highly ligand-specific proteins whose reactivity includes a binding step.
DNA DETERMINATION
Residual host cell DNA is a potential process-specific impurity in a biotechnology-derived product. The residual DNA is unique for each product because it is dependent on the host organism and the process recovery procedure used to manufacture the product. Although adverse health effects have not been reported from biologicals because of their DNA content, regulatory agencies have requested manufacturers to ensure that the DNA level in biotechnology-derived products is reduced to low levels.
The technique of DNA hybridization (dot blot analysis) is the most sensitive, routine DNA assay available to determine the DNA content of products. It is valuable as a purification process assay to demonstrate that a low level of DNA has been attained early in the manufacturing process. The method relies on the hybridization of cellular DNA from the sample with either specific
32P-labeled or chemically modified DNA probes obtained from the DNA of the host cell. The analysis is performed by first isolating any residual DNA in the sample by a procedure that may include hydrolysis of the protein, chromatography, organic extractions, and alcohol precipitation. The isolated DNA is denatured and then applied to a nitrocellulose or nylon membrane along with a set of serially-diluted host DNA standards. Positive and negative DNA controls are also applied and the membrane may be baked at approximately 80
or placed under UV light to complete binding of the DNA to the membrane. A DNA probe is then prepared either by nick translation, random primer synthesis, or chemical modification of a DNA extract of the host cell. The DNA probe is purified and thermally denatured at 95
. It is then added to the baked or UV-treated membrane and allowed to hybridize with the DNA of the samples at approximately 42
in the presence of formamide or at higher temperatures without formamide for 24 to 48 hours. The membrane is subsequently placed between two X-ray films and exposed to produce an autoradiograph or is developed by immunochemical means using an enzyme conjugate/substrate system similar to ELISA and/or Western blot. The DNA of the sample is estimated by visual comparison of the dot intensity of the sample to those of the diluted DNA standards. The autoradiogram can also be scanned by optical densitometry. The sensitivity of the assay, i.e., 10 to 250 pg, is determined by the limit of visual detection above background of the serially-diluted DNA standards.
Other methods for DNA determination have been developed using biosensor technology. This methodology currently determines total DNA/nucleic acid impurities rather than specific host cell DNA. This technology may become quite valuable in the future, especially when more specific DNA binding methods are developed. Finally, the recently developed polymerase chain reaction (PCR) technology, which involves DNA amplification, may prove useful in detection and identification of contaminant DNA. Quantitative use of this technology, however, will require further development.
CARBOHYDRATE DETERMINATION
One of the possible post-translational modifications that occurs on proteins is the covalent attachment of oligosaccharide chains. Glycosylation is a characteristic of recombinant proteins that are expressed from eukaryotic cell lines. Although the polypeptide chain of a glycoprotein is synthesized under the direct control of the genetic code, oligosaccharides are not primary gene products, but are synthesized by enzymes known as glycosyltransferases. This synthesis results in microheterogeneity of the carbohydrate chains. Also, glycosylation is cell-line dependent, so glycoproteins with identical polypeptide chains made in different cell lines may have considerably different carbohydrate structures. The sugars commonly found in glycoproteins include neutral sugars (D-galactose, D-mannose, and L-fucose), amino sugars (N-acetylglucosamine and N-acetylgalactosamine), and the acidic sugar, sialic acid.
Two main approaches can be taken to determine the sugars covalently attached to the glycoprotein. Both are based on the understanding that microheterogeneity is a common phenomenon among glycoproteins, and that the information represents either average composition or representative structures.
The first approach is the determination of the composition of sugars in a glycoprotein, which can be performed by several methods. Neutral sugars and sialic acid may be determined by simple colorimetric tests. Total neutral sugars can be determined following reaction with phenol and sulfuric acid and measuring the absorbance of the solution at about 490 nm compared to a standard curve. Following mild acid hydrolysis and periodate oxidation, free sialic acid content can be determined with thiobarbituric acid and the absorbance of the solution at about 550 nm compared to a standard curve. Individual neutral sugars can be determined following acid hydrolysis by several methods. Underivatized, they can be separated by HPIEC at high pH and quantitated by pulsed amperometric detection. They may also be converted to the alditol peracetates with acetic anhydride or to the aldononitrile acetates with hydroxylamine hydrochloride and pyridine prior to peracetylation, and the derivatives separated by gas chromatography.
The second approach in determining the carbohydrate composition is to release and separate individual oligosaccharide structures covalently attached to the glycoprotein. This requires an understanding of the types of structures attached. The attachment of sugars to proteins can occur in two major ways: through an O-glycosidic bond involving the hydroxyl group of serine, threonine, or modified amino acids such as hydroxylysine or hydroxyproline, or through the N-glycosidic bond of asparagine. O-linked oligosaccharides can be released from the protein following beta-elimination under alkaline conditions and reduction of the reducing end sugar with sodium borohydride. N-linked oligosaccharides can be released chemically by hydrazinolysis or enzymatically by one of a variety of specific glycosidases, such as endo H, endo F, or peptido-N-glycanase. The oligosaccharides can then be separated by HPIEC at high pH and quantitated with pulsed amperometric detection. This results in an oligosaccharide or carbohydrate map analogous to the peptide map for the protein.
ADVENTITIOUS and ENDOGENOUS AGENT DETECTION
Specific assays pertaining to biotechnology focus on the detection of bacteria, fungi, mycoplasma, and viruses. These reflect the possible contaminants that may occur in both bacterial fermentation and mammalian cell culture. Control is exerted in a variety of ways including characterization of the master seed bank and the working cell banks to ensure freedom from these contaminants, evaluation of raw materials, the design and operation of closed manufacturing systems, testing of production lots, and validation of specific manufacturing processes to ensure that contaminants would be inactivated or removed if present.
Freedom of final sterile dosage forms from bacteria and fungi is usually evaluated by tests for sterility as described in
Sterility Tests 71. Mycoplasma assays are performed by standard cultivation methods employing aerobic and anaerobic incubation of solid medium in plates and semisolid broth in tubes and must comply with the code of federal regulations (21 CFR 610.12). In addition, noncultivable mycoplasma are detected microscopically by using the Hoechst bisbenzimide staining method.
Various methods that are used for the detection of adventitious virus contamination in cell lines include inoculation of indicator cell lines selected for their ability to support the replication of a broad range of viruses and monitoring these for markers of virus infection such as cytopathology, hemadsorption, hemagglutination, and immunofluorescence; inoculation of intact animals and monitoring for illness and death; inoculation of animals and, after four weeks, collection and evaluation of serum for antibodies to specific viruses of concern; and specific immunologic assays or genetic probes for some viruses of concern that cannot be detected by the other methods listed.
The expression of endogenous retrovirus genes is highly variable among different mammalian cells and cell lines. The unpredictable nature of their expression and the diversity of their biochemical and biological properties preclude the use of a single test and instead require an integrated testing strategy. Test methods generally used include transmission electron microscopy of cells from the master seed bank and ultracentrifuged pellets of cell-free, cell culture harvests; various assays for infectious retroviruses that use retrovirus-susceptible indicator cell lines; reverse transcriptase activity; and induction of retroviruses in cells of the master cell bank with chemicals known to induce retroviruses. In addition to classical virological methods, newer techniques such as molecular probe hybridization are also beginning to be used for these evaluations.